Правила коагуляции электролитами. Порог коагуляции. Правило Шульце-Гарди. Виды коагуляции: концентрационная и нейтрализационная. Коагуляция смесями электролитов. Явление "неправильные ряды". Механизм и кинетика коагуляции
Коагуляцией
называется процесс слипания частиц с образованием крупных агрегатов. В результате коагуляции система теряет свою седиментационную устойчивость, так как частицы становятся слишком крупными и не могут участвовать в броуновском движении.
Коагуляция является самопроизвольным процессом, так как она приводит к уменьшению межфазной поверхности и, следовательно, к уменьшению свободной поверхностной энергии.
Различают две стадии коагуляции.
1 стадия – скрытая коагуляция. На этой стадии частицы укрупняются, но еще не теряют своей седиментационной устойчивости.
2 стадия - явная коагуляция. На этой стадии частицы теряют свою седиментационную устойчивость. Если плотность частиц больше плотности дисперсионной среды, образуется осадок.
Причины коагуляции многообразны. Едва ли существует какое либо внешнее воздействие, которое при достаточной интенсивности не вызывало бы коагуляцию.
Правила коагуляции:
1. Все сильные электролиты, добавленные к золю в достаточном количестве, вызывают его коагуляцию.
Минимальная концентрация электролита, при которой начинается коагуляция, называется порогом коагуляции
Ck
.
Иногда вместо порога коагуляции используют величину VK
, называемую коагулирующей способностью. Это объем золя, который коагулирует под действием 1 моля электролита:
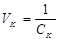 , ,
т.е. чем меньше порог коагуляции, тем больше коагулирующая способность электролита.
2. Коагулирующим действием обладает не весь электролит, а только тот ион, заряд которого совпадает по знаку с зарядом противоиона мицеллы лиофобного золя. Этот ион называют ионом–коагулянтом
.
3. Коагулирующая способность иона–коагулянта тем больше, чем больше заряд иона.
Количественно эта закономерность описывается эмпирическим правилом Щульце – Гарди:
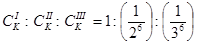 или или  . .
где a - постоянная для данной системы величина;
Z
– заряд иона – коагулянта;
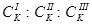 - порог коагуляции однозарядного, двухзарядного, трехзарядного иона-коагулянта. - порог коагуляции однозарядного, двухзарядного, трехзарядного иона-коагулянта.
Правило устанавливает
, что коагулирующие силы иона тем больше, чем больше его валентность
. Экспериментально установлено, что ионы с высшей валентностью имеют значение порога коагуляции ниже, чем ионы с низшей. Следовательно, для коагуляции лучше брать ионы с высшей степенью окисления. Если валентность ионов одинакова, то коагулирующая способность зависит от размеров и степени гидратации ионов. Чем больше радиус иона, тем больше его коагулирующая способность. По этому правилу составлены лиотропные ряды
. Органические ионы-коагулянты, как правило, лучше коагулируют гидрозоли, чем неорганические, т.к. они легко поляризуются и адсорбируются. С точки зрения двойного электрического слоя (ДЭС) считается, что коагуляция идет в том случае, когда z-потенциал > 30 мВ.
Коагулирующая способность иона при одинаковом заряде тем больше, чем больше его кристаллический радиус
. Причина с одной стороны, в большой поляризуемости ионов наибольшего радиуса, следовательно, в их способности притягиваться поверхностью, состоящей из ионов и полярных молекул. С другой стороны, чем больше радиус иона, тем меньше, при одной и той же величине заряда, гидратация иона. Гидратная же оболочка уменьшает электрическое взаимодействие. Коагулирующая способность органических ионов больше по сравнению с неорганическими ионами.
Для однозарядных неорганических катионов коагулирующая способность убывает в следующем порядке:
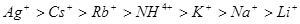 - лиотропный ряд. - лиотропный ряд.
При увеличении концентрации иона–коагулянта z – потенциал мицеллы золя уменьшается по абсолютной величине. Коагуляция может начинаться уже тогда, когда z – потенциал снижается до 0,025 – 0, 040 В (а не до нуля).
При коагуляции золя электролитами различают концентрационную
и нейтрализационную
коагуляцию.
Концентрационная коагуляция
имеет место, когда она происходит под действием индифферентного электролита вследствие сжатия диффузного слоя противоионов и уменьшения абсолютного значения z-потенциала.
Рассмотрим концентрационную коагуляцию золя хлорида серебра, стабилизированного нитратом серебра, при введении в золь нитрата калия.
Формула мицеллы имеет вид:
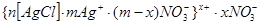 . .
На рис. 3.1.2.1 показан график изменения потенциала в ДЭС мицеллы хлорида серебра. Кривая 1 относится к исходной мицелле, кривая 2 – после добавления KNO
3
в количестве, вызывающем коагуляцию. При добавлении KNO
3
диффузный слой противоионов сжимается, формула мицеллы приобретает вид:
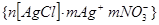

На рис. 3.1.2.2 представлены потенциальные кривые, характеризующие взаимодействие частиц в этом золе. z-потенциал исходной коллоидной частицы положительный, это создаёт потенциальный барьер коагуляции ∆
U
к
=0
(кривая 2 рис. 3.1.2.2). Поэтому ничто не мешает коллоидным частицам сблизиться на такое расстояние, где преобладают силы притяжения – происходит коагуляция. Так как в данном случае причиной коагуляции является увеличение концентрации противоионов, она называется концентрационной коагуляцией.
Для этого случая теория дает формулу
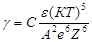
где g - порог коагуляции;
С
– константа, слабо зависящая от асимметрии электролита, т.е. отношение числа зарядов катиона и аниона;
А
– константа;
е
– заряд электрона;
e - диэлектрическая проницаемость;
Z
– заряд коагулирующего иона;
Т
– температура.
Из уравнения следует, что значение порогов коагуляции для одно-, двух-, трех-, четырех- зарядных ионов должны соотноситься 1 к (1/2)6
к (1/3)6
к (1/4)6
и т.д., т.е. обосновывается ранее представленное эмпирическое правило Шульце – Гарди.
Нейтрализационная коагуляция
происходит при добавлении к золю неиндифферентного электролита. При этом потенциалопределяющие ионы связываются в малорастворимое соединение, что приводит к уменьшению абсолютных величин термодинамического потенциала, а следовательно, и z-потенциала вплоть до нуля.
Если взять в качестве исходного только что рассмотренный золь хлорида серебра, то для нейтрализации потенциалопределяющих ионов Ag+
в золь необходимо ввести, например, хлорид калия. После добавления определённого количества этого неиндифферентного электролита мицелла будет иметь вид:

В системе не будет ионов, способных адсорбироваться на поверхности частицы AgCl
, и поверхность станет электронейтральной. При столкновении таких частиц происходит коагуляция.
Так как причиной коагуляции в данном случае является нейтрализация
потенциалопределяющих ионов, такую коагуляцию называют нейтрализационной коагуляцией.
Необходимо отметить, что для полной нейтрализационной коагуляции неиндифферентный электролит должен быть добавлен в строго эквивалентном количестве.
При коагуляции смесью электролитов различают два типа процессов:
· гомокоагуляция
· гетерокоагуляция
Гомокоагуляция
- укрупнение подобных частиц в больший агрегат осадка. Причем в процессе отстаивания мелкие частицы растворяются, а крупные увеличиваются за их счет. На этом основано явление активации и перекристаллизации. Этот процесс описывается уравнением Кельвина – Томсона:
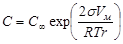 , ,
где С
¥
- растворимость макрочастиц;
С
– растворимость микрочастиц;
V
м
– молярный объем;
R
– универсальная газовая постоянная;
T
– температура;
r
– радиус частиц.
Из уравнения следует, что концентрация вокруг маленького радиуса больше, поэтому диффузия идет от бóльшей концентрации к меньшей.
При втором типе происходит слияние разнородных частиц или прилипание частиц дисперсной системы на вводимые в систему чужеродные тела или поверхности.
Гетерокоагуляция
- взаимная коагуляция разнородных дисперсных систем.
Коагуляция смесью электролитов имеет большое практическое значение, так как даже при добавлении к золю одного электролита-коагулянта, в действительности коагуляция происходит, по крайней мере, под влиянием двух электролитов, так как в системе содержится электролит-стабилизатор. Кроме того, в технике для коагуляции часто применяют смесь двух электролитов. Понимание закономерностей взаимного действия электролитов важно также при исследовании воздействия биологически активных ионов на органы и ткани живого организма.
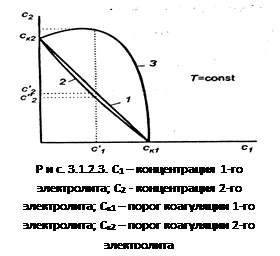
При коагуляции
золя смесью двух и более электролитов
возможны три случая (рис. 3.1.2.3). По оси абсцисс отложена концентрация первого электролита С
1
, а C
к1
– его порог коагуляции. Аналогично по оси ординат отложена концентрация второго электролита С
2
, а С
к2
– его порог коагуляции.
1.
Аддитивное действие электролитов
(линия 1 рис. 3.1.2.3). Электролиты действуют как бы независимо один от другого, их суммарное действие складывается из воздействий каждого из электролитов. Если с1
´
- концентрация первого электролита, то для коагуляции золя концентрация второго электролита должна быть равной с2
´
. Аддитивность наблюдается обычно при сходстве коагулирующей способности обоих электролитов.
2. Синергизм действия
(линия 2 рис. 3.1.2.3). Электролиты как бы способствуют друг другу – для коагуляции их требуется меньше, чем нужно по правилу аддитивности (с2
″
< c2
′
). Условия, при которых наблюдается синергизм, сформулировать трудно.
3. Антагонизм действия
(линия 3 рис. 3.1.2.3). Электролиты как бы противодействуют друг другу и для коагуляции их следует добавить больше, чем требуется по правилу аддитивности. Антагонизм наблюдается при большом различии в коагулирующем действии электролитов.
Существует несколько теорий, объясняющих явление антагонизма. Одной из его причин может служить химическое взаимодействие между ионами.
Например, для золя AgCl, стабилизированного хлоридом калия, коагулирующем действием обладают катионы. Например, большой коагулирующей способностью обладает четырёхзарядный ион тория Th4+
. Однако если взять для коагуляции смесь Th(NO3
)4
и K2
SO4
, то коагулирующая способность этой смеси значительно меньше, чем отдельно взятого Th(NO3
)4
. Связано это с тем, что в результате химической реакции образуется комплекс:
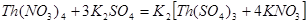
и вместо четырёхзарядных ионов Th
4+
в золе будут находиться однозарядные катионы K
+
, коагулирующее действие которых значительно слабее (правило Шульце-Гарди).
Гетероадагуляция
- прилипание частиц дисперсной фазы к вводимой в систему чужеродной поверхности.
Одной из причин этого явления является адсорбция стабилизатора на этой поверхности. Например: отложение коллоидных частиц на волокнах при крашении и дроблении.
Для гидрофобных золей в качестве ВМС обычно используют белки, углеводы, пектины; для неводных золей – каучуки.
При введении в коллоидный раствор электролитов, содержащих многовалентные ионы с зарядом противоположные заряду частиц, наблюдается явление "неправильные ряды"
. Оно состоит в том, что при добавлении к отдельным порциям золя все возрастающего его количества электролита золь сначала остается устойчивым, затем в определенном интервале концентраций происходит коагуляция; далее золь снова становится устойчивым и, наконец, при повышении концентрации электролита опять наступает коагуляция уже окончательная. Подобные явления могут вызывать и большие органические ионы. Объясняется это тем, что при весьма малых количествах введенного электролита ионов недостаточно, чтобы коагулировать золь, т. е. значение x- потенциала остается выше привычного (рис. 3.1.2.4). При больших количествах электролита его ионы проявляют коагулирующее действие. Этот интервал концентраций отвечает значениям x- потенциала частиц от x критического первого знака до x критического другого знака.

При еще больших концентрациях многовалентные ионы перезаряжают коллоидную частицу и золь опять устойчивый. В этой зоне x-потенциал опять выше критического значения, но обратен по знаку частицам исходного золя. Наконец, при высоком содержании исходного электролита многовалентные ионы снова снижают значение x-потенциала ниже критического и снова происходит окончательная коагуляция.
Повышение агрегативной устойчивости золя путём введения в него высокомолекулярного соединения (ВМС) называется коллоидной защитой.
Происходит образование защитной пленки на поверхности золя (гидратной или ВМС), препятствующей взаимодействию частиц электролита.
В качестве количественной характеристики коагуляции Зигмонди предложил использовать скорость коагуляции
.
Скорость коагуляции
u
- это изменение концентрации коллоидных частиц в единицу времени при постоянном объеме системы
.
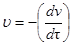 , ,
где n - концентрация частиц;
t
- время.
Знак "-" стоит потому, что концентрация частиц со временем уменьшается, а скорость всегда положительна.
Степень коагуляции a:
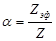 , ,
где Z
- общее число столкновений частиц в единицу времени; Z
эф
- число эффективных столкновений (т.е. столкновений, приводящих к коагуляции) в единицу времени.
Если a = 0, коагуляция не происходит, коллоидный раствор агрегативно устойчив.
Если a = 1, происходит быстрая коагуляция, т.е. каждое столкновение частиц приводит к их слипанию.
Если 0 <a< 1, наблюдается медленная коагуляция, т.е. только некоторые столкновения частиц приводят к их слипанию.
Чтобы частицы при столкновении слиплись, а не разлетелись как упругие шары, должен быть преодолен потенциальный барьер коагуляции Δ
U
к
. Следовательно, коагуляция произойдет только в том случае, когда коллоидные частицы будут обладать кинетической энергией, достаточной дл преодоления этого барьера. Для увеличения степени коагуляции необходимо снижать потенциальный барьер. Это может быть достигнуто добавлением к золю электролита – коагулянта.
Зависимость скорости коагуляции от концентрации электролита представлена на рис. 3.1.2.5.
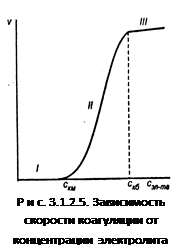
На графике видны три участка:
I. 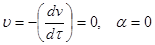 . .
Следовательно, кинетическая энергия kТ
<< Δ
U
к
, (k
– постоянная Больцмана) – лиофобный золь агрегативно устойчив.
II. 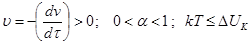 , т.е. потенциальный барьер коагуляции больше, но соизмерим с кинетической энергией коллоидных частиц, причем с увеличением концентрации электролита – коагулянта он уменьшается, а скорость коагуляции возрастает. Скм
– порог медленной коагуляции, Скб
– порог быстрой коагуляции. Этот участок кривой выражает зависимость: , т.е. потенциальный барьер коагуляции больше, но соизмерим с кинетической энергией коллоидных частиц, причем с увеличением концентрации электролита – коагулянта он уменьшается, а скорость коагуляции возрастает. Скм
– порог медленной коагуляции, Скб
– порог быстрой коагуляции. Этот участок кривой выражает зависимость:
На этом участке происходит медленная коагуляция.
III. 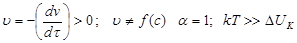
Каждое столкновение приводит к слипанию частиц – идет быстрая коагуляция.
Теория быстрой коагуляции
, разработанная М. Смолуховским в 1916 г., основана на следующих положениях.
1. Рассматриваемая система является монодисперсной, радиус частиц r
.
2. 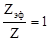 , т.е. все столкновения являются эффективными. , т.е. все столкновения являются эффективными.
3. Рассматриваются только столкновения первичных частиц.
4. Кинетика коагуляции подобна кинетике бимолекулярной реакции:
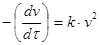 , ,
где k
– константа скорости коагуляции.
Проинтегрируем это уравнение, разделив переменные:
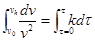
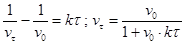 , ,
где u
0
– концентрация частиц золя в начальный момент времени;
u
t
– концентрация частиц золя в момент времени t
.
Для характеристики быстрой коагуляции используется период коагуляции(период половинной коагуляции)
q
.
Период коагуляции (
q
) – это время, через которое концентрация коллоидных частиц уменьшается в два раза
.
При 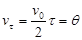

Согласно теории быстрой коагуляции, константа коагуляции зависит от коэффициента диффузии и может быть вычислена по уравнению
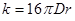
Если подставить в это уравнение величину коэффициента диффузии, получим:
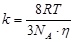
Таким образом, зная вязкость дисперсионной среды и температуру, можно вычислить константу скорости быстрой коагуляции. Теория Смолуховского неоднократно проверялась экспериментально и получила блестящее подтверждение, несмотря на сделанные автором допущения.
Медленная коагуляция
связана с неполной эффективностью столкновений вследствие существования энергетического барьера. Простое введение величины степени коагуляции a в формулы теории Смолуховского не привело к согласию теории с опытом. Более совершенную теорию медленной коагуляции разработал Н.Фукс. Он ввел в кинетическое уравнение коагуляции множитель, учитывающий энергетический барьер коагуляции Δ
U
к
:
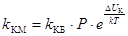 , ,
где k
КМ
– константа скорости медленной коагуляции;
k
КБ
- константа скорости быстрой коагуляции;
Р
– стерический фактор;
ΔU
к
- потенциальный барьер коагуляции;
k
– постоянная Больцмана.
Таким образом, для расчета константы скорости медленной коагуляции необходимо знать потенциальный барьер коагуляции, величина которого зависит прежде всего от z– потенциала.
Фактор устойчивости
, или коэффициент замедления
W
, показывает, во сколько раз константа скорости медленной коагуляции меньше константы скорости быстрой коагуляции.
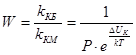 , ,
Следует отметить пять факторов устойчивости, среди которых два первых играют главную роль.
1. Электростатический фактор устойчивости.
Он обусловлен наличием ДЭС и x– потенциала на поверхности частиц дисперсной фазы.
2. Адсорбционно – сольватный фактор устойчивости.
Он обусловлен снижением поверхностного натяжения в результате взаимодействия дисперсионной среды с частицей дисперсной фазы. Этот фактор играет заметную роль, когда в качестве стабилизаторов используются коллоидные ПАВ.
3. Структурно – механический фактор устойчивости.
Он обусловлен тем, что на поверхности частиц дисперсной фазы образуются пленки, обладающие упругостью и механической прочностью, разрушение которых требует времени и затраты энергии. Этот фактор устойчивости реализуется в тех случаях,
когда в качестве стабилизаторов используются высокомолекулярные соединения (ВМС).
4. Энтропийный фактор устойчивости.
Коагуляция приводит к уменьшению числа частиц в системе, следовательно, к уменьшению энтропии (ΔS
<0), а это приводит к увеличению свободной энергии системы ΔG
>0. Поэтому система самопроизвольно стремится оттолкнуть частицы друг от друга и равномерно (хаотично) распределить по объему системы. Этим обусловлен энтропийный фактор устойчивости. Однако число частиц в коллоидном растворе по сравнению с истинным раствором такой же массовой концентрации гораздо меньше, поэтому роль энтропийного фактора невелика. Но если частицы стабилизированы веществами, обладающими длинными гибкими цепями (ВМС) и потому имеющими много конформаций, то при сближении таких частиц их защитные слои вступают во взаимодействие. Это взаимодействие непременно приводит к уменьшению числа возможных конформаций, а значит – к уменьшению энтропии. Поэтому система стремится оттолкнуть частицы друг от друга.
5. Гидродинамический фактор устойчивости.
Ему способствует увеличение плотности и динамической вязкости дисперсионной среды.
В реальных системах действуют, как правило, несколько факторов устойчивости. Каждому фактору соответствует специфический способ его нейтрализации. Это затрудняет создание общей теории устойчивости. Пока существуют лишь частные теории.
|