ОПРЕДЕЛЕНИЕ УРОНОВЫХ КИСЛОТ И ПОЛИУРОНИДОВ
Казань, 2009
Введение
В состав кислых полисахаридов и полиуронидов древесины входят звенья двух гексуроновых кислот — D-глюкуроновой и D-галактуроновой. Звенья D-глюкуроновой кислоты преимущественно содержатся в полисахаридах гемицеллюлоз, а звенья D-галактуроновой кислоты в виде галактуронанов входят в состав пектиновых веществ.
Методы определения общего содержания уроновых кислот в древесине основаны на реакции декарбоксилирования под действием сильных минеральных кислот. Одновременно происходит гидролиз полисахаридов. Обычно используют 12...19%-ную соляную кислоту. Звенья уроновых кислот при этом превращаются в звенья пентоз, из которых в результате реакций гидролиза гликозидных связей и дегидратации образуется фурфурол.
Выход фурфурола составляет только 35...40% от теоретического, что, вероятно, объясняется образованием из глюкуроноксиланов при гидролизе сначалаальдобиуроновой кислоты, в которой уроновая кислота связана смолекулой ксилозы. Альдобиуроновые кислоты гидролизуютсядальше с образованием составляющих Сахаровуже значительно труднее.
Дальнейший анализ сводится к определению образовавшегося диоксида углерода гравиметрическими или титриметриче-скими методами. Во всех методах диоксид углерода поглощают щелочами и по количеству поглощенного диоксида углерода рассчитывают массовую долю уроновых кислот в древесине.
Для количественного определения CO2
предложены различные методы: поглощение CO2
аскаритом или же раствором гидроксида калия в приборах для поглощения газов с последующим гравиметрическим определением — взвешиванием прибора-поглотителя до и после опыта; поглощение CO2
растворами гидроксида бария или натрия с последующим титрованием избытка щелочи и др. Для анализа разработан ряд методов, в том числе микро- и полумикрометодов, и конструкций установок. Из сферы реакции CO2
удаляют с помощью азота или воздуха, очищенного от CO2
.
Обработку древесины соляной кислотой обычно проводят в реакционной колбе, нагреваемой масляной или глицериновой баней, при температуре 140...145°С.
При проведении реакции декарбоксилирования следует учитывать возможное присутствие в исследуемом материале карбонатов. С этой целью рекомендуют проводить предварительное нагревание реакционной смеси при температуре бани 70°С в течение 10...15 мин.
Реакция декарбоксилирования уроновых кислот 12...13%-ным раствором HCI обычно заканчивается через 4 ч. Применение 19%-ного раствора HCI снижает необходимую продолжительность обработки, но одновременно приводит к некоторому увеличению выхода диоксида углерода из других источников, помимо уроновых кислот. Опытным путем установили, что для декарбоксилирования уроновых кислот в этих условиях требуется примерно 200 мин, для компенсации ошибок из-за постороннего CO2
рекомендуют обработку продолжительностью 140...160 мин.
Все методы определения выхода CO2
из древесины и другого растительного сырья, основанные на реакции декарбоксилирования, обычно дают завышенные результаты, так как диоксид углерода частично образуется из гексоз при их разложении в кислой среде, причем железо,, переходящее при этом из древесины в гидролизат, служит катализатором этой реакции. Выход диоксида углерода в результате разложения гексоз может достигать 0,2....0,3% от абсолютно сухой древесины. Следовательно, получение при анализе результатов в этих пределах не обязательно служит доказательством присутствия уроновых кислот. Однако установить точную поправку практически невозможно, и при малом содержании уроновых кислот метод становится неточным.
Зная общую массовую долю уроновых кислот в древесине, найденную по реакции декарбоксилирования, и долю уроновых кислот, входящих в виде полигалактуронанов р состав пектиновых кислот, по разности можно рассчитать массовую долю звеньев уроновых кислот, входящих в состав полисахаридов гемицеллюлоз.
Для раздельного определения в растительном сырье глюкуроновой и галактуроновой кислот используют гидролиз растительной ткани с последующим анализом гидролизатов методами бумажной хроматографии, электрофореза на бумаге или газожидкостной хроматографии.
При определении звеньев уроновых кислот в технической и окисленной целлюлозах учитывают поправку на образование диоксида углерода из самой целлюлозы. Кинетика выделения CO2
из звеньев уроновых кислот и самой целлюлозы различна, что и позволяет отличить одну реакцию от другой. Из уроновых кислот CO2
образуется с большой первоначальной скоростью, которая постепенно снижается до нуля. Из целлюлозы же CO2
образуется медленно, сохраняя постоянную скорость реакции на протяжении длительного времени. Таким образом, форма кривой скорости реакции может служить признаком присутствия или отсутствия групп уроновых кислот в целлюлозе. Уистлер и Невел разработали метод и прибор для определения уроновых кислот в целлюлозе с гравиметрическим определением CO2
после поглощения его аскаритом. Для внесения поправки на образование CO2
из целлюлозы в аналогичных условиях проводят обработку чистой хлопковой целлюлозы.
1. Определение уроновых кислот полумикрометодом Беркера
Метод основан на обработке древесины 19%-ной соляной кислотой с поглощением CO2
раствором гидроксида натрия и последующим титрованием его избытка соляной кислотой. Для того чтобы карбонат натрия не мешал установлению точки эквивалентности, добавляют раствор хлорида бария

Анализ проводят в стеклянном аппарате, состоящем из двугорлой остродонной колбы / вместимостью 50 см3
, снабженной капилляром 2, доходящим до дна колбы, и обратным шариковым холодильником 3. В верхней части холодильника имеется ловушка 4, заполненная гранулированным цинком. Ловушка соединена с поглотителем 5. Все части аппарата соединены шлифами и стянуты пружинами. Во время работы аппарата через него пропускают из газометра ток азота или воздуха, освобожденного от диоксида углерода. Скорость потока газа через капилляр регулируют краном и дополнительным зажимом, установленным на резиновом шланге, соединяющем капилляр с газометром через систему очистки газа.
Методика анализа. Навеску воздушно-сухих опилок массой 0,2...0,3 г помещают в реакционную колбу и добавляют пипеткой 3 см3
19%-ного раствора HCI. Через установку в течение 10 мин пропускают ток азота для удаления CO2
из установки. Затем в поглотитель пипеткой вливают 5 см3
раствора гидроксида натрия концентрацией 0,25 моля/дм3
, присоединяют поглотитель и устанавливают ток азота со скоростью один пузырек в 2...3 с.
Под реакционную колбу ставят предварительно нагретую до 145°С баню со сплавом Вуда, причем уровень сплава должен быть примерно на 2,5 мм ниже уровня жидкости в колбе. Нагревание колбы при температуре бани 145°С продолжают 2,5 ч. Затем баню убирают и еще 10 мин пропускают азот со скоростью 2...3 пузырька в секунду. После этого поглотитель отсоединяют, содержимое его выливают в коническую колбу вместимостью 100 см с притертой пробкой, тщательно смывают остатки раствора дистиллированной водой, присоединяя промывную воду к раствору в колбе. Добавляют в колбу 2 см3
10%-ного раствора BaCb и 2 капли раствора фенолфталеина. Оттитровывают избыток щелочи соляной кислотой с концентрацией 0,1 моль/дм3
до обесцвечивания фенолфталеина. Аналогично проводится контрольный опыт. Массовую долю уроновых кислот,% к абсолютно сухой древесине, рассчитывают по формуле

где а — расход на титрование раствора HCl концентрацией 0,1 моль/дм3
в контрольном опыте, см3
; b— расход на титрование раствора HCl концентрацией 0,1 моль/дм3
в рабочем'опыте, см3
; 0,0097 — масса уроновых кислот, соответствующая 1 см3
раствора HCI концентрацией 0,1 моль/дм3
, г; g— масса абсолютно сухой навески древесины, г.
Расхождение между результатами двух параллельных определений не должно превышать 0,5%.
2. Пектиновые вещества и методы их определения
В древесине в срединной пластинке пектиновые вещества присутствуют преимущественно в нерастворимой форме, т. е. в виде протопектина. В состав комплекса протопектина входят галактуронаны, арабинаны и галактаны, частично связанные между собой химическими связями. Нерастворимость протопектина в воде обусловлена образованием карбоксильными группами галактуронанов солей кальция и магния, с возникновением поперечных мостиков между цепями. Поэтому для извлечения пектиновых веществ из древесины вместо экстрагирования водой применяют обработку горячими растворами оксалата или цитрата аммония, приводящую к разрушению этих мостиков в результате обменной реакции с образованием нерастворимых солей — оксалатов или цитратов кальция и магния.
Для количественного определения полиуронидной части пектиновых веществ можно использовать гравиметрический кальций-пектатный метод, основанный на гидролизе сложноэфирных групп пектиновой кислоты под действием гидроксида натрия, в результате чего пектиновая кислота превращается в полигалактуроновую кислоту, называемую пектовой кислотой, с последующим осаждением из раствора пектата кальция.
Кальций-пектатный метод имеет ряд недостатков: неполное осаждение полиуроновых кислот при одновременном осаждении нейтральных полисахаридов; невозможность определения всех составляющих комплекса пектиновых веществ, т. е. не только его полиуронидной, но и нейтральной части; большая длительность анализа.
Для одновременного количественного определения массовой доли в древесине и состава пектиновых веществ предложен спектрофотометрический метод, позволяющий с помощью о-толуидинового реагента определять полиуроновые кислоты, гексозавы и пентозаны непосредственно в растворе без осаждения. Метод основан на извлечении пектиновых веществ раствором цитрата аммония с последующим гидролизом полиуронидов и полисахаридов серной кислотой и определением образовавшихся моносахаридов и галактуроновой кислоты с о-толуидиновым реагентом. Образующиеся окрашенные соединения определяют спектрофотометрический методом.
В работе установлены оптимальные условия анализа, которые и приводятся в изложенной ниже методике. Для нахождения моносахаридного состава нейтральных полисахаридов комплекса пектиновых веществ можно использовать хроматографический анализ.
3. Определение пектиновых веществ спектрофотометрический методом с отолуидиновым реагентом
Методика анализа. Навеску измельченной древесины или другого растительного сырья массой около 1 г помещают в грушевидную колбу вместимостью 100 см3
, добавляют 40 см3
1%-ного раствора цитрата аммония и кипятят с обратным холодильником на электрической плитке в течение I ч. Полученный раствор отфильтровывают горячим через конусообразную стеклянную воронку с бумажным фильтром в мерную колбу на 100 см3
. Попавшие на фильтр частицы древесины смывают новой порцией 1%-ного раствора цитрата аммония и повторяют обработку. Остаток древесины в колбе и на фильтре промывают небольшим объемом горячей дистиллированной воды и присоединяют промывные воды к раствору в мерной колбе. После охлаждения колбы с раствором до 20°С его объем доводят до метки дистиллированной водой.
Для проведения гидролиза полиуронидов и полисахаридов отбирают пипеткой 5 см3
раствора пектиновых веществ в кругло-донную колбу вместимостью 25 см3
, добавляют пипеткой 5 см3
раствора серной кислоты концентрацией 4 моль/дм3
, вносят стеклянные «кипятильники» и кипятят с обратным холодильником на электрической плитке в течение 3 ч.
Гидролизат охлаждают до комнатной температуры и нейтрализуют равным объемом раствора карбоната натрия концентрацией 2 моль/дм3
. Нейтрализацию из-за сильного вспенивания проводят в высокой пробирке. Можно нейтрализовать не весь, а только часть гидролизата, достаточную для последующего анализа.
Для проведения анализа углеводных компонентов в пробирку с конусом типа П4 вместимостью 10 см3
пипеткой вносят 0,5 см3
нейтрализованного гидролизата, добавляют пипеткой 4 см3
о-толуидинового реагента и перемешивают содержимое. Параллельно в другой пробирке готовят раствор сравнения. Обе пробирки нагревают на кипящей водяной бане в течение 30 мин. Затем пробирки охлаждают до комнатной температуры, переливают растворы в кюветы с толщиной слоя 1 см3
и на спектрофотометре измеряют оптические плотности раствора при аналитических длинах волн 365, 385 и 630 нм, используя в качестве раствора сравнения смесь воды и OTP.
В такой же кювете измеряют оптическую плотность контрольного раствора — нейтрализованного гидролизата, разбавленного в 9 раз для учета разбавления при анализе с OTPпри длинах волн 365 и 385 нм, используя в качестве раствора сравнения дистиллированную воду.
Для расчета коэффициентов поглощения строят градуировочные графики для используемого спектрофотометра по растворам чистых Сахаров
Вывод уравнений и расчет коэффициентов. В применении к анализируемому раствору Сахаров уравнения Фирордта будут иметь вид

где D36
Sи D385
— оптические плотности анализируемого раствора
при длинах волн 365 и 385 нм за вычетом поглощения гекеоз; с, и 'Ci—концентрации соответственно пентоз и галактуроновой кислоты, мкг/см3
; — коэффициенты поглощения продуктов реакции с OTP пентоз и галактуроновой кислоты при длинах волн 365 и 385 нм; d— толщина кюветы, см, d= 1 см. — коэффициенты поглощения продуктов реакции с OTP пентоз и галактуроновой кислоты при длинах волн 365 и 385 нм; d— толщина кюветы, см, d= 1 см.
Решая систему двух уравнений, получают уравнения для расчета концентрации, мкг/см3
, пентоз
C = ZiD385
-SD365
и галактуроновой кислоты
2=CD365
— EDis
5
,
где соответствующие коэффициенты будут равны
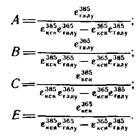
Расчет массовой доли
пектиновых веществ.
После расчета концентрацией, мкг/см3
, галактуроновой кислоты Ci
, пентоз Cl
и гекеоз рассчитывают массы полисахаридов и полиуронидов с учетом общего объема раствора пектиновых веществ, разбавленийзпри гидролизе и нейтрализации и коэффициентов пересчета моносахаридов в полисахариды:
Галактуронан = сгму
· 400 · 0,907 · IO-
Y
Пентозаны = сп<
.нтоз
·
400 · 0,88 · 10"6
г,
Гексозаны = сгексоэ
·
400 · 0,90 ·
IO
-
V
Затем рассчитывают общую массу пектиновых веществ как сумму масс всех полисахаридов и полиуронидов тпв
= галактуронан + пентозаны -4- гексозаны.
Массовую долю пектиновых веществ Xnii
,% к исходной абсолютно сухой древесине, рассчитывают по формуле

где тпа
— масса пектиновых веществ, г; g— масса абсолютно сухой навески обессмоленной древесины, г; K3
— коэффициент экстрагирования органическим растворителем.
Построение градуировочных графиков. Готовят по пять растворов чистых Сахаров концентрацией 40, 80, 120, 160 и 200 мкг/см3
. Каждый раствор обрабатывают OTP в условиях определения пектиновых веществ и измеряют оптические плотности при 365 и 385 нм, а для растворов глюкозы — дополнительно при 630 нм и строят соответствующие графики, откладывая по оси абсцисс концентрации растворов, мкг/см3
, а по оси ординат — значения оптической плотности D.
|