Контрольная работа №1
1 Приведите проекционные формулы оптических изомеров соединений

Определите, число изомеров и укажите, какие из них являются энантиомерами, а какие – диастереомерами.
Решение
а). Число изомеров – 2, оба изомера являются по отношению друг к другу энантиомерами.
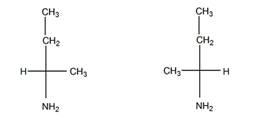
б). Число изомеров – 2, оба - энантиомеры
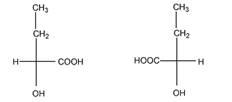
в). Число изомеров – 4.

Пары энантиомеров: I и III, II и IV; пары диастереомеров: I и II, III и IV.
2
Приведите механизм реакции радикального замещения
(
SR
)
на примере бромирования 2-метилпропана и циклогексана. Объясните устойчивость третичного радикала по сравнению с вторичным и первичным
Решение
Механизм бромирования 2-метилпропана
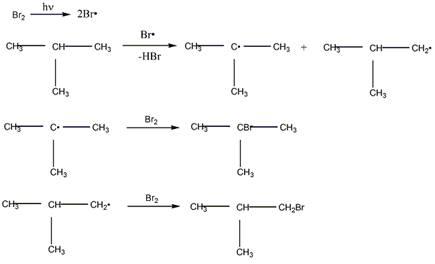
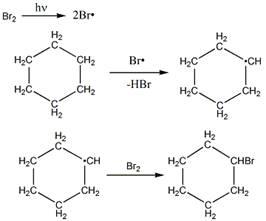
Механизм бромирования циклогексанаость свободных радикалов определяется энергией их образования из алканов. Энергия, необходимая для образования различных типов радикалов, уменьшается в следующем порядке: СН3 > первичный > вторичный > третичный.
Если для образования одного радикала требуется меньше энергии, чем для образования другого, то это может означать только то, что в сравнении с образующимся алканом один радикал содержит меньше энергии и более устойчив, чем другой (см. рисунок ниже):

Абсолютное содержание энергии, например, метильного и этильного радикалов не сравнивается; просто говорят, что различие в энергиях между метаном и метильным радикалом больше, чем между этаном и этильным радикалом.
3 Дайте определение понятию «кислотности» органических соединений по Бренстеду-Лоури и расположите в ряд по возрастанию кислотных свойств следующие соединения: фенол, пропантиол-1, пропиловый спирт, пропановая кислота, пропан, пропанамин-1.
Укажите вид и знак электронных эффектов заместителей. Обоснуйте кислотные свойства указанных веществ, исходя из стабильности соответствующих анионов
Решение
По теории Брёнстеда (протолитической теории) кислотность и основность соединений связывается с переносом протона Н. Кислота <-> Н + Основание. Кислота и основание образуют сопряженную кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряженное ей основание, и напротив, чем сильнее основание, тем слабее сопряженная ему кислота. Например, хлороводородная кислота сильнее, чем уксусная кислота и соответственно ацетат-ион будет более сильным основанием, чем хлорид-ион. Кислоты Брёнстеда (протонные кислоты) нейтральные молекулы или ионы, способные отдавать протон (доноры протонов). Основания Брёнстеда — нейтральные молекулы или ноны, способные присоединить протон (акцепторы протонов). Кислотность и основность являются не абсолютными, а относительными свойствами соединений: кислотные свойства обнаруживаются лишь в присутствии основания; основные свойства только в присутствии кислоты.
Большинство органических соединений можно рассматривать как кислоты, поскольку в них содержатся поляризованные связи атома водорода с различными элементами (О, N, S, С). Органические кислоты классифицируют по природе кислотного центра:
ОН-кислоты: спирты, фенолы, карбоновые кислоты, сульфокислоты, гидроксикислоты, аминокислоты;
· SH-кислоты: тиоспирты, SH-содержащие аминокислоты и др. соединения;
· NH-кислоты: амины, имины, гетероциклические соединения с атомом азота;
· СН-кислоты: углеводороды, радикалы гетерофункциональных соединений.
Для количественной
характеристики кислотных свойств используется величина
pKa
= - lgKa
,
где Ка
– константа кислотности. Чем меньше рКа
, тем больше кислотность по Бренстеду.
Качественной
характеристикой кислотных свойств может служить стабильность образующегося аниона. Сила кислоты определяется стабильностью аниона, образующегося из этой кислоты: чем стабильнее анион, тем сильнее кислота. Стабильность аниона, в свою очередь, определяется характером распределения отрицательного заряда аниона и зависит от ряда факторов:
1) природы атома в кислотном центре (электроотрицательности и поляризуемости элемента);
2) характера связанного с кислотным центром органического радикала (электроноакцепторного или электронодонорного);
3) сольватационных эффектов.
Электроотрицательность
имеет значение, когда сравнивается кислотность соединений, имеющих одинаковые радикалы и элементы кислотного центра, относящиеся к одному и тому же периоду периодической системы Д.И. Менделеева (т.е. когда практически не изменяется поляризуемость). Чем более электроотрицательным является элемент в кислотном центре, тем он более способен нести отрицательный заряд, и тем стабильнее образующийся анион, и соответственно, сильнее кислота.
С—Н
кислота
|
N—H
кислота
|
О—Н
кислота
|
S—H
кислота
|
О—Н
кислоты
|
С2
Н5
СH2
←Н
пропан
|
 Н Н
С3
Н7
N←H
пропанамин
|
С3
Н7
О←Н
Пропиловый спирт
|
С2
Н5
S←H
Пропантиол-1
|
С6
Н5
О←Н
фенол
|
С2
Н5
СОО←Н
уксусная кислота
|
| рКа
=50 |
рКа
≈ 30 |
рКа
≈ 18 |
рКа
≈ 12 |
рКа
= 10 |
рКа
= 4,9 |
Кислотность соединений в ряду слева направо увеличивается.
У пропана, пропанамина и пропилового спирта кислотность, ввиду отсутствия электроноакцепторных групп у кислотообразующей частицы обеспечивается исключительно электроотрицательностью этой самой частицы.
В пределах группы таблицы элементов Менделеева стабильность анионов возрастает с увеличением атомного номера элемента, так как увеличивается объем электронных орбиталей, и создается лучшая возможность для делокализации отрицательного заряда. Поэтому пропантиол является более сильной кислотой, чем пропанол.

Фенильная группа (бензольное кольцо) обладает слабым отрицательным индуктивным эффектом и делокализует образовавшийся на атоме кислорода отрицательный заряд по всему бензольному кольцу:

Аналогично, для пропановой кислоты отрицательный заряд делокализуется по системе сопряженных связей:

4 Объясните, как изменяется основность в указанном ряду соединений. Как практически можно подтвердить основность самого сильного основания этого ряда?
n
-Хлоранилин, метиламин, метилпропиламин, п- нитроанилин, анилин, дифениламин
Решение
На основность аминов влияют различные факторы: электронные эффекты углеводородных радикалов, пространственное экранирование радикалами атома азота, а также способность образующихся ионов к стабилизации за счет сольватации в среде растворителя. В результате +I-эффекта алкильных групп основность алифатических аминов в газовой фазе (без растворителя) растет в ряду: первичные < вторичные < третичные. Однако в растворах оснoвные свойства третичных аминов проявляются слабее, чем у вторичных и даже первичных аминов, так как три радикала создают пространственные препятствия для сольватации образующихся аммониевых ионов. По этой же причине основность первичных и вторичных аминов снижается с увеличением размеров и разветвленности радикалов.
Основность ароматических аминов зависит также от характера заместителей в бензольном кольце. Электроноакцепторные заместители (-F, -Cl, -NO2
и т.п.), а также фенильная группа, уменьшают основные свойства ариламина по сравнению с анилином, а электронодонорные (алкил, -OCH3
, -N(CH3
)2
и др.), напротив, увеличивают.
Основность аминов (в растворе) возрастает в ряду:
дифениламин < анилин<п-хлоранилин <п-нитроанилин < метиламин < метилпропиламин
Водные растворы алифатических аминов проявляют щелочную реакцию, т.к. при их взаимодействии с водой образуются гидроксиды алкиламмония, аналогичные гидроксиду аммония:

5 Установите строение углеводорода С5
Н12
, при монобромировании которого образуется третичный галогеноалкан. Искомый углеводород нельзя получить по реакции Вюрца без побочных продуктов. Запишите уравнения реакций получения этого углеводорода гидрированием алкена и щелочной плавкой соли карбоновой кислоты
Решение
Строение искомого углеводорода представлено ниже

Гидрирование алкена

Щелочная плавка соли карбоновой кислоты

6 Углеводород С6
Н]2
присоединяет 1 моль Вг2
, растворяется в холодной концентрированной серной кислоте, при гидрировании превращается в 2-метилпентан, а при окислении перманганатом калия в кислой среде при нагревании образует среди продуктов реакции уксусную кислоту. Предложите его структурную формулу. Напишите уравнения перечисленных реакций
Решение
Строение углеводорода

Присоединение брома

Растворение в холодной концентрированной серной кислоте

Гидрирование

Окисление перманганатом калия

7 Установите строение углеводорода С6
Н10
, если известно, что он обесцвечивает бромную воду, образует красный осадок с аммиачным раствором оксида меди (I), а в результате присоединения воды в присутствии сульфата ртути превращается в изобутилметилкетон
Решение

8 Определите, какие продукты будут преимущественно образовываться при бромировании (в присутствии катализатора
FeBr
3
)
следующих соединений

Решение
а). 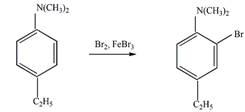
б). 
в). 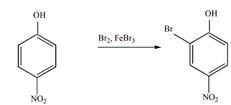
г).
9 Определите структурную формулу углеводорода состава С9
Н10
, который: а) обесцвечивает реактив Вагнера; б) вступает в реакцию полимеризации; в) существует в виде цис-транс-изомеров;
г) при окислении даёт бензойную кислоту
Решение
Структурная формула углеводорода (бета-метилстирол)

а). обесцвечивание реактива Вагнера
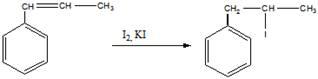
б). полимеризация бета-метилстирола (с добавлением перекиси бензоила в качестве инициатора)

в). Цис-транс-изомерия

транс-изомер цис-изомер
г). окисление

10 Установите структурную формулу соединения С5
Н11
Вг, легко вступающего в реакцию гидролиза, протекающую по механизму
SN
1.
Продукт гидролиза при дегидратации и последующем озонолизе даёт смесь уксусного альдегида и ацетона
Решение
Структурная формула вещества и последующие реакции приведены ниже

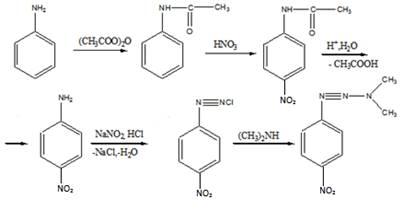
11 Напишите, какие соединения получатся в результате последовательного действия на анилин уксусного ангидрида, нитрующей смеси, воды (в присутствии НС1),
NaNO
2
(
в
присутствии Н
Cl
), N,N-диметиланилина
Решение
12 Углеводороды состава:
a
)
C
8
H
6
и б) С9
Н8
обесцвечивают бромную воду, при окислении образуют бензойную кислоту, с аммиачным раствором нитрата серебра дают осадок. Напишите структурные формулы этих углеводородов
Решение
Строение соединений приведено на рисунке

а) фенилацетилен б) 1-фенилэтин
13. Приведите уравнения реакций по следующим схемам:
а) Бензол —» нитробензол —» анилин —> ацетанилид —> n -нитроацетанилид —»
п-нитроанилин;
б) Хлоробензол —»м - бромобензойная кислота
Решение
а).
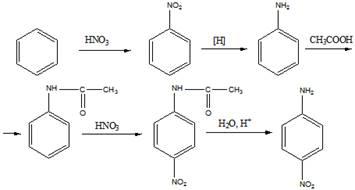
б).
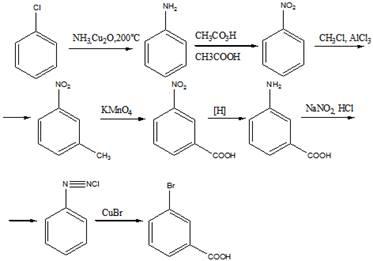
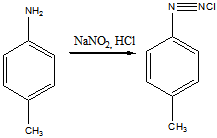
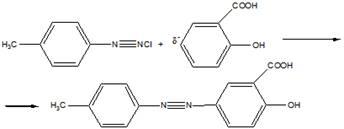
14 Напишите схему получения азокрасителя, используя в качестве диазо и азосоставляющих соответственно
n
-толуидин и салициловую кислоту. Укажите
условия реакции. Опишите механизмы реакций диазотирования и азосочетания
Решение
Для получения диазосоставляющего компонента п-толуидин подвергают реакции диазотирования.
Затем идет стадия азосочетания:
Механизм диазотирования следующий:
В водном растворе сильной неорганической кислоты азотистая кислота, образующаяся как малодиссоциирующее соединение в результате взаимодействия нитрита натрия и соляной кислоты, частично протонируется с образованием нитрозацидий-катиона :

Нитрозацидий-катион очень активный электрофильный агент. Согласно кинетическим данным, этот катион в водном растворе гораздо быстрее реагирует с неорганическими анионами, присутствующими в растворе, чем с ароматическим амином.

В результате образуются новые реагенты: азотистый ангидрид, хлористый или бромистый нитрозил, которые могут быть электрофильными агентами при диазотировании в разбавленном водном растворе.
Диазотированию подвергается амин в виде свобoдного основания. Лимитирующей стадией всего процесса диазотирования является образование N-арилнитрозоаммония, как это предполагал Е. Бамбергер еще в 1900 году, далее следует ряд быстрых протолитических равновесий, приводящих к диазосоединению, как к конечному продукту.

Механизм азосочетания:
Азосочетание включает две стадии-присоединение катиона диазония к азосоставляющей и отщепление протона:

15 Установите структурную формулу первитина
C
10
H
15
N
,
относящегося к важному классу лекарственных веществ, возбуждающих нервную систему, снимающих усталость и повышающих работоспособность. Это соединение обладает следующими свойствами: а) имеет асимметрический атом углерода (не в бензильном положении); б) взаимодействует с минеральными кислотами с образованием солей; в) не даёт изонитрильную реакцию, но ацилируется уксусным ангидридом; г) при окислении превращается в бензойную кислоту
Строение первитина:
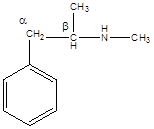
а). Асимметрический атом углерода находится в бета-положении от бензольного кольца, так как окружен 4-мя разными заместителями.
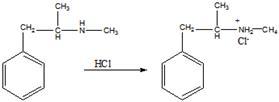
б). Взаимодействие с соляной кислотой.
в). Ацилирование уксусным ангидридом

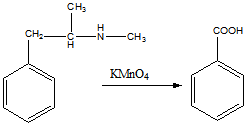
г). Окисление перманганатом калия
Контрольная работа №2 Вариант 1
1 Получите несколькими способами изобутиловый спирт. Напишите для него уравнения реакций с:
a
)
Na
;
б)
CH
3
MgI
;
в) НС1. Укажите, в каких реакциях спирт проявляет кислотные свойства, в каких основные. Назовите продукты реакций. Приведите механизм для случая (в)
Решение
Получение изопропилового спирта:
Гидролиз хлоризобутана

Оксосинтез из пропилена в присутствии НСо(СО)4
при 120-160°С и 20-35 МПа:

Количество изобутилового спирта, получаемого оксосинтезом, на 1 тонну пропилена 305-320 кг.
Восстановление изобутаналя

Реакции:
а). Взаимодействие с металлическим натрием. Спирт проявляет кислотные свойства.

Продукт – изобутилалкоголят натрия
б).Взаимодействие с реактивом Гриньяра. Спирт здесь проявляет основные свойства.

Продукт – 2-метилбутан
в).Взаимодействие с соляной кислотой. Спирт проявляет основные свойства

Продукт – хлоризобутан
2. Осуществите превращения, все продукты назовите, укажите условия химических превращений.

Опишите механизм получения вещества В.

Решение
а).
Реакция А:

Продукты: хлороформ и изобутановая кислота
Реакция Б:

Продукт: оксим метилизопропилкетона
Реакция В:

Продукт реакции В: 2-иодизопропилметилкетон
Механизм образования продукта В:

б).Приведенная ниже цепочка реакций проходит в безводном эфире (англ. Ester)
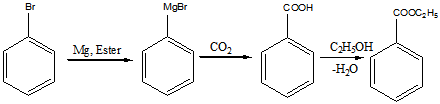
Реакция F:

Продукты: бензойная кислота и этанол
Реакция G:

Продукты: бензоат натрия и этанол.
3 Определите строение вещества состава С2
НС
l
3
O
, которое оказывает успокаивающее и гипнотическое действие и обладает следующими свойствами: а) реагирует с гидросульфитом натрия и гидроксиламином; б) реагируя с водой, даёт кристаллический продукт; в) при щелочном расщеплении образует хлороформ и формиат натрия
(
HCOONa
)
Ответ: Хлораль

а). Реакция с гидросульфатом натрия

Реакция с гидроксиламином

б). реакция с водой

в). щелочное расщепление

4 Оптически активный спирт С5
Н12
O
при дегидратации превращается в соединение, озонолиз которого дает ацетон и уксусный альдегид. Установите строение исходного спирта. Какова конфигурация спирта, если он с уксусной кислотой в присутствии минеральной кислоты образует сложный эфир с
d
-
конфигурацией
Решение

При этерификации конфигурация асимметричного атома углерода не изменяется, так что исходный спирт D-ориентирован.
5.1 Аминокислоты, полипептиды, белки
Аминокислоты - класс азотсодержащих органических кислот, имеющих общие черты строения, которые могут быть представлены общей формулой
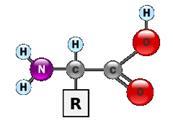
Аминокислоты отличаются друг от друга типом аминокислотного остатка R. Таким образом молекула каждой аминокислоты содержит специфическую часть (боковую группу - R) и неспецифическую часть. Существует около 20 различных аминокислот. Аминокислоты являются строительными блоками (мономерами), из которых строятся все белковые молекулы (полимеры). Основные 20 аминокислот : аланин (ала, ala, A) аргинин (арг, arg, R), aспарагин (асн, asn, N), аспартат (асп, asp, D), валин (вал, val, V), гистидин (гис, his, H), глицин (гли, gly, G), глутамат (глу, glu, E),. глутамин (глн, gln, Q) изолейцин , (илей,ile, I), лейцин , (лей, leu, L), лизин , (лиз, lys, K), метионин , (мет, met, M), пролин , (про, pro, P), серин (сер, ser, S), тирозин , (тир, tyr, Y), треонин , (тре, thr, T), триптофан (три, trp, W), фенилаланин (фен, phe, F), цистеин (цис, cys, C). Свободные аминокислоты составляют примерно 0.5% от веса клетки , входящие в состав белков - около 15%..
Аминокислоты - структурные элементы, из которых построены белки. Представляют собою карбоновые кислоты, содержащие одну или две аминогруппы. Общим признаком аминокислот, входящих в состав белка (исключение составляет пролин), является наличие свободной карбоксильной группы и свободной незамещенной аминогруппы у альфа-углеродного атома.Наиболее рациональная классификация аминокислот основана на различиях в полярности R-групп. R-группы подразделяются на четыре основных класса:
1) неполярные, или гидрофобные ;
2) полярные, но незаряженные ;
3) положительно заряженные ;
4) отрицательно заряженные (при pH 6-7) .
Полипептиды. или просто пептиды, природные или синтетич. соед., молекулы к-рых построены из остатков a-аминокислот, соединенных между собой пептидными (амидными) связями C(O) NH. Могут содержать в молекуле также неаминокислотную компоненту (напр., остаток углевода). По числу аминокислотных остатков, входящих в молекулы пептидов, различают ди-пептиды, трипептиды, тетрапептиды и т.д. Пептиды, содержащие до 10 аминокислотных остатков, наз. олигопептидами, содержащие более 10 аминокислотных остатков полипептидами Природные полипептиды с мол. массой более 6 тыс. называются белками.
Белки́ (протеи́ны, полипепти́ды) — высокомолекулярные органические вещества, состоящие из соединённых в цепочку пептидной связью аминокислот. В живых организмах аминокислотный состав белков определяется генетическим кодом, при синтезе в большинстве случаев используется 20 стандартных аминокислот. Множество их комбинаций дают большое разнообразие свойств молекул белков. Кроме того, аминокислоты в составе белка часто подвергаются посттрансляционным модификациям, которые могут возникать и до того, как белок начинает выполнять свою функцию, и во время его «работы» в клетке. Часто в живых организмах несколько молекул белков образуют сложные комплексы, например, фотосинтетический комплекс.
Кроме последовательности аминокислот полипептида (первичной структуры), крайне важна трёхмерная структура белка, которая формируется в процессе фолдинга (от англ. folding, «сворачивание»). Трёхмерная структура формируется в результате взаимодействия структур более низких уровней. Выделяют четыре уровня структуры белка[15]:
Первичная структура — последовательность аминокислот в полипептидной цепи. Важными особенностями первичной структуры являются консервативные мотивы — сочетания аминокислот, важных для функции белка. Консервативные мотивы сохраняются в процессе эволюции видов, по ним часто удаётся предсказать функцию неизвестного белка.
Вторичная структура — локальное упорядочивание фрагмента полипептидной цепи, стабилизированное водородными связями и гидрофобными взаимодействиями. Ниже приведены некоторые распространённые типы вторичной структуры белков:
α-спирали — плотные витки вокруг длинной оси молекулы, один виток составляют 3,6 аминокислотных остатка, и шаг спирали составляет 0,54 нм[16] (так что на один аминокислотный остаток приходится 0,15 нм), спираль стабилизирована водородными связями между H и O пептидных групп, отстоящих друг от друга на 4 звена. Спираль построена исключительно из одного типа стереоизомеров аминокислот (L). Хотя она может быть как левозакрученной, так и правозакрученной, в белках преобладает правозакрученная. Спираль нарушают электростатические взаимодействия глутаминовой кислоты, лизина, аргинина. Расположенные близко друг к другу остатки аспарагина, серина, треонина и лейцина могут стерически мешать образованию спирали, остатки пролина вызывает изгиб цепи и также нарушает α-спирали.
β-листы (складчатые слои) — несколько зигзагообразных полипептидных цепей, в которых водородные связи образуются между относительно удалёнными друг от друга (0,347 нм на аминокислотный остаток[16]) в первичной структуре аминокислотами или разными цепями белка, а не близко расположенными, как имеет место в α-спирали. Эти цепи обычно направлены N-концами в противоположные стороны (антипараллельная ориентация). Для образования β-листов важны небольшие размеры боковых групп аминокислот, преобладают обычно глицин и аланин.
π-спирали;
310-спирали;
неупорядоченные фрагменты.
Третичная или трёхмерная структура — пространственное строение полипептидной цепи (набор пространственных координат составляющих белок атомов). Структурно состоит из элементов вторичной структуры, стабилизированных различными типами взаимодействий. В стабилизации третичной структуры принимают участие:
ковалентные связи (между двумя остатками цистеина — дисульфидные мостики);
ионные связи между противоположно заряженными боковыми группами аминокислотных остатков;
водородные связи;
гидрофильно-гидрофобные взаимодействия.
При взаимодействии с окружающими молекулами воды белковая молекула «стремится» свернуться так, чтобы неполярные боковые группы аминокислот оказались изолированы от водного раствора; на поверхности молекулы оказываются полярные гидрофильные боковые группы.
Четверичная структура (или субъединичная, доменная) — взаимное расположение нескольких полипептидных цепей в составе единого белкового комплекса. Белковые молекулы, входящие в состав белка с четвертичной структурой, образуются на рибосомах по отдельности и лишь после окончания синтеза образуют общую надмолекулярную структуру (можно считать её и молекулой, если между разными полипептидными цепями, как это нередко бывает, образуются дисульфидные мостики). В состав белка с четвертичной структурой могут входить как идентичные, так и различающиеся полипептидные цепочки. В стабилизации четвертичной структуры принимают участие те же типы взаимодействий, что и в стабилизации третичной. Надмолекулярные белковые комплексы могут состоять из десятков молекул, многие из них сравнимы по размеру с рибосомами и в последние годы часто описываются как органоиды (см., напр., протеасома). Нередко в их состав входят молекулы РНК (см., напр., сплайсосома).
5.2 Углеводы (моносахариды, полисахариды). Гликозиды. Крахмал (амилоза, амилопектин), декстран (полиглюкин)
Углево́ды (сахариды) — общее название обширного класса природных органических соединений. Название происходит от слов «уголь» и «вода». Причиной этого является то, что первые из известных науке углеводов описывались брутто-формулой Cx
(H2
O)y
, формально являясь соединениями углерода и воды.
С точки зрения химии углеводы являются органическими веществами, содержащими неразветвленную цепь из нескольких атомов углерода, карбонильную группу, а также несколько гидроксильных групп.
Моносахариды (от греческого monos: единственный, sacchar: сахар), — органические соединения, одна из основных групп углеводов; самая простая форма сахара; являются обычно бесцветными, растворимыми в воде, прозрачными твердыми веществами. Некоторые моносахариды обладают сладким вкусом. Моносахариды — стандартные блоки, из которых синтезируются дисахариды (такие, как сахароза) и полисахариды (такие, как целлюлоза и крахмал), содержат гидроксильные группы и альдегидную (альдозы) или кетогруппу (кетозы). Каждый углеродный атом, с которым соединена гидроксильная группа (за исключением первого и последнего) является хиральным, давая начало многим изомерным формам. Например, галактоза и глюкоза — альдогексозы, но имеют различные химические и физические свойства. Моносахариды, как и все углеводы, содержат только 3 элемента (C,O,H).
К моносахаридам относятся:
Глюко́за («виноградный сахар», декстроза) встречается в соке многих фруктов и ягод, в том числе и винограда, отчего и произошло название этого вида сахара. Является шестиатомным сахаром (гексозой).
Фруктоза, или плодовый сахар — моносахарид, который в свободном виде присутствует почти во всех сладких ягодах и плодах. Многие предпочитают заменять сахар не синтетическими препаратами, а природной фруктозой.
Галактоза — один из простых сахаров. Отличается от глюкозы пространственным расположением водородной и гидроксильной групп у 4-го углеродного атома. Содержится в животных и растительных организмах, в том числе в некоторых микроорганизмах. Входит в состав молочного сахара. При окислении образует галактоновую, галактуроновую и слизевую кислоты. Хорошо растворима в воде
Полисахари́ды — общее название класса сложных высокомолекулярных углеводов, молекулы которых состоят из десятков, сотен или тысяч мономеров — моносахаридов.
Полисахариды необходимы для жизнедеятельности животных и растительных организмов. Они являются одним из основных источников энергии, образующейся в результате обмена веществ организма. Они принимают участие в иммунных процессах, обеспечивают сцепление клеток в тканях, являются основной массой органического вещества в биосфере.
Была установлена многообразная биологическая активность полисахаридов растительного происхождения: антибиотическая, противовирусная, противоопухолевая, антидотная[источник не указан 322 дня]. Полисахариды растительного происхождения выполняют большую роль в уменьшении липемии и атероматоза сосудов благодаря способности давать комплексы с белками и липо-протеидами плазмы крови.[1]
К полисахаридам относятся, в частности:
декстрин — полисахарид, продукт гидролиза крахмала;
крахмал — основной полисахарид, откладываемый, как энергетический запас у растительных организмов;
гликоген — полисахарид, откладываемый, как энергетический запас в клетках животных организмов, но встречается в малых количествах и в тканях растений;
целлюлоза — основной структурный полисахарид клеточных стенок растений;
галактоманнаны — запасные полисахариды некоторых растений семейства бобовых, такие как гуаран и камедь рожкового дерева;
глюкоманнан — полисахарид, получаемый из клубней конняку, состоит из чередующихся звеньев глюкозы и маннозы, растворимое пищевое волокно, уменьшающее аппетит;
амилоид — применяется при производстве пергаментной бумаги.
Гликози́ды — органические соединения, молекулы которых состоят из двух частей: углеводного (пиранозидного или фуранозидного) остатка и неуглеводного фрагмента (т. н. агликона). В качестве гликозидов в более общем смысле могут рассматриваться и углеводы, состоящие из двух или более моносахаридных остатков. Преимущественно кристаллические, реже аморфные вещества, хорошо растворимые в воде и спирте.
Гликозиды представляют собой обширную группу органических веществ, встречающихся в растительном (реже в животном) мире и/или получаемых синтетическим путём. При кислотном, щелочном, ферментативном гидролизе они расщепляются на два или несколько компонентов — агликон и углевод (или несколько углеводов). Многие из гликозидов токсичны или обладают сильным физиологическим действием, например гликозиды наперстянки, строфанта и другие.
Крахма́л — полисахариды амилозы и амилопектина, мономером которых является альфа-глюкоза. Крахмал, синтезируемый разными растениями под действием света (фотосинтез) имеет несколько различных составов и структуру зёрен.
Амилоза (от греч. ámylon — крахмал) — один из основных полисахаридов крахмала, состоящий из линейных или слаборазветвлённых цепочек молекул глюкозы, соединённых связями между 1-м и 4-м углеродными атомами.

Ами́лопекти́н (от греч. ámylon — крахмал, pēktes — сбитый, сплочённый) — один из основных полисахаридов крахмала, состоящий из разветвленных цепочек молекул глюкозы, соединённых связями как между 1-м и 4-м, так и 1-м и 6-м углеродными атомами.

Декстри́н — полисахарид, получаемый термической обработкой картофельного или кукурузного крахмала. Образуется из крахмала в ротовой полости человека под действием α-амилаз.

5.3 Пектиновые вещества. Эфиры целлюлозы метил-, карбоксиметил- и натрийкарбоксиэтилцеллюлоза). Растительные камеди
Пекти́новые вещества́ (от греч. pektos — свернувшийся, замёрзший) — полисахариды, образованные остатками главным образом галактуроновой кислоты. Присутствуют во всех наземных растениях (особенно много в плодах) и в некоторых водорослях. Способствуют поддержанию в тканях тургор, повышают засухоустойчивость растений, устойчивость овощей и плодов при хранении. Используются в пищевой и фармацевтической промышленности как студнеобразующие вещества. Получают пектиновые вещества из яблочных выжимок, жома сахарной свёклы и т. п.
Эфиры целлюлозы, производные целлюлозы общей формулы [C6
H7
O2
(OH)3-х
(OR)х
]n
, где n - степень полимеризации; x - число групп ОН, замещенных в одном звене макромолекулы целлюлозы (степень замещения - СЗ); R - алкил, ацил или остаток минер, кислоты. Каждое звено макромолекулы содержит 3 группы ОН, которые способны вступать в реакции с образованием простых и сложных эфиров; в случае смешанных эфиры целлюлозы э. замещающие радикалы различны.
Наиболее распространены эфиры целлюлозы э.: простые - карбоксиметилцеллюлоза, метилцеллюлоза, этилцеллюлоза, а также метилгидроксипропилцеллюлоза, оксипропилцеллюлоза, цианэтилцеллюлоза; сложные - целлюлозы ацетаты, целлюлозы нитраты, а также ацетилфталилцеллюлоза, ацетопропионаты, ацетобутираты и сульфаты целлюлозы. Упомянутые эфиры целлюлозы э. производят во многие странах десятками и сотнями тысяч т в год.
Св-ва эфиров целлюлозы э. зависят главным образом от числа и, СЗ и типа заместителя R. Так, степень полимеризации (в среднем 150-500) значительно влияет преимущественно на прочностные и вязкостные свойства эфиры целлюлозы э., обеспечивая их пригодность для переработки. СЗ определяет их физических-механические и химический свойства. Средняя СЗ лежит в пределах 0-3; однако чаще СЗ рассчитывают не на одно, а на 100 элементарных звеньев макромолекул целлюлозы и обозначают (например, для триацетилцеллюлозы= 280-290). Регулируют СЗ изменением условий синтеза: концентрации алкилирующего или этерифицирующего агента, температуры, продолжительности и др.
Растворимость эфиры целлюлозы э. зависит от содержания и соотношения заместителей и свободный групп ОН. Например, ацетат целлюлозы, имеющий СЗ 0,5-0,8 и 1,5-1,8, раств. соответственно в воде и смеси ацетон - вода (7:3); ацетат целлюлозы со СЗ 2,2-2,6 растворим в ацетоне и метилцеллозольве, со СЗ > 2,6 - в метиленхлориде и смеси метиленхлорид - этанол (9:1). При увеличении длины цепи алкильного радикала гидрофобность эфиры целлюлозы э. повышается и они способны растворим в неполярных растворителях (например, бутил- и пропилцеллюлоза уже нерастворимы в воде и растворим в органических растворителях). Вообще растворимость эфиры целлюлозы э. в органических растворителях возрастает с повышением температуры и уменьшается с увеличением молекулярной массы.
С увеличением в заместителе числа атомов С для всех эфиры целлюлозы э. уменьшаются влагопоглощение, температуры размягчения и плавления. Сложные эфиры термически нестабильны и обладают низкой химический стойкостью к действию кислот и щелочей. Простые эфиры устойчивы в кислотах и щелочах и выдерживают нагревание до сравнительно высоких температур, не разлагаясь и не выделяя свободный кислот, вызывающих коррозию металлов. Сложные и некоторые простые эфиры целлюлозы э.- хорошие диэлектрики.
Для производства эфиры целлюлозы э. используют облагороженную хлопковую и древесную (сульфатную и сульфитную) целлюлозу. Выбор ее вида определяется областью применения того или иного эфира. Для повышения скорости и равномерности О-алкилирования и однородности эфиры целлюлозы э. независимо от способа их получения исходную целлюлозу обязательно предварительно активируют. В производстве простых эфиров целлюлозу обрабатывают раствором NaOH, в результате чего она набухает и приобретает повышенную реакционную способность (щелочная целлюлоза) вследствие облегчения диффузии компонентов этерифицирующей смеси внутрь материала. В производстве сложных эфиров целлюлозу обрабатывают уксусной или др. кислотой при повышенной температуре в парах либо растворами этих кислот. Обычно, чем выше температура активации, тем меньше ее продолжительность.
Простые эфиры целлюлозы э. получают в автоклавах при повышенной температуре взаимодействие щелочной целлюлозы с алкилхлоридами и (или) 3-и 4-членными гетероциклический соединение, напр, этилен- и пропиленоксидами, сультонами (пром. способы), диалкилсульфатами (лабораторная способ), непредельными соединение с двойными связями (например, акрилонитрил, акриламид). Так, О-алкилированием щелочной целлюлозы монохлоруксусной кислотой получают Na-соль карбоксиметилцеллюлозы, диэтиламиноэтилхлоридом -диэтиламиноэтилцеллюлозу, акрилонитрилом - цианэтилцеллюлозу, этилен- и пропиленоксидами - гидроксиэтил- и гидроксипропилцеллюлозы. Образование простых эфиров катализируется основаниями и всегда сопровождается побочными реакциями.
Сложные эфиры целлюлозы э. в промышлености получают:
1. Этерификацией целлюлозы кислородсодержащими не-органическое и карбоновыми (например, НСООН) кислотами. Этим способом получают нитраты, сульфаты и формиаты целлюлозы. Этерификация ее Н3РО4 в смеси с мочевиной дает фосфаты целлюлозы. Вследствие обратимости реакции применяют конц. кислоты и водоотнимающие добавки.
2. Действием на целлюлозу преимущественно ангидридов кислот в среде органическое растворителей или разбавителей в присутствии катализаторов (в основные минеральных кислот). Таким способом получают эфиры на основе карбоновых кислот жирного ряда С2 - С4 (например, ацетаты целлюлозы). Действием смесей ангидридов различные кислот или кислоты и ангидрида др. кислоты производят смешанные эфиры целлюлозы э. (например, ацетопропионаты и ацетобутираты целлюлозы).
Лабораторная способы получения сложных эфиров: действие на целлюлозу изоцианатов (Ц. э. карбаминовой кислоты - замещенные уретаны, карбанилаты целлюлозы); переэтерификация (бораты, фосфаты, стеарат целлюлозы). При синтезе эфиры целлюлозы э. в кислой среде побочные продукты почти не образуются.
Области применения сложных, а также простых и смешанных эфиры целлюлозы э. весьма разнообразны. Осн. направления использования: производство искусств. волокон (см. Ацетатные волокна, Вискозные волокна, Гидратцеллюлозные волокна, Медноаммиачные волокна); эфироцеллюлозных пластмасс (см. Этролы); различные пленок, полупроницаемых мембран (см. Пленки полимерные, Фотографические материалы); лакокрасочных материалов (см. Грунтовки, Лакокрасочные покрытия, Шпатлевки, Эфироцеллюлозные лаки). Ц. э. применяют также как загустители, пластификаторы и стабилизаторы глинистых растворов для буровых скважин, асбо- и гипсоцементных штукатурных смесей, обмазочных масс для сварных электродов, водоэмульсионных красок, красителей (при печати по тканям), зубных паст, парфюмерно-косметич. средств, водно-жировых фармацевтич. составов, пищевая продуктов (например, соков, муссов); связующие в литейных производствах; эмульгаторы при полимеризации; ресорбенты загрязнений в синтетич. моющих средствах; флотореагенты при обогащении различные руд; текстиль-но-вспомогат. вещества (например, аппретирующие и шлихтующие); компоненты клеевых композиций и др.
Растительные камеди — вещества, выделяющиеся в виде прозрачных густеющих масс при повреждении растений (при механическом их поранении или при патологических процессах, вызываемых бактериями или грибками). Из выделенной растением аморфной массы можно извлечь камеди действием щелочи с последующим осаждением кислотой. Это — гидрофильные вещества, в большинстве случаев хорошо растворимые в воде с образованием клейких растворов.
Камеди представляют собой нейтральные соли (кальциевые, магниевые, калиевые) высокомолекулярных кислот, состоящих из остатков гексоз, пентоз, метилпентоз и уроновых кислот. Из гексоз все камеди содержат D-галактозу (некоторые, кроме того, еще D-маннозу), из пентоз — L-арабинозу (некоторые, кроме того, ксилозу). Метилпентоза — рамноза, или фукоза, — содержится не во всех камедях. Уроновая кислота всех камедей, кроме камеди трагаканта, — это D-глюкуроновая кислота; камедь трагаканта содержит D-галактуроновую кислоту.
При нагревании камедей на водяной бане, иногда с разбавленными кислотами, т. е. в мягких условиях, происходит их «аутогидролиз», заключающийся в отщеплении молекул моносахаридов и олигосахаридов. Изучение строения камедей весьма осложнено трудностями получения их в чистом виде. Наиболее изучена аравийская камедь.
Аравийская камедь, или гуммиарабик (кальциевая соль арабовой кислоты), получается из сенегальской акации и имеет применение, в частности, в медицине. При полном кислотном гидролизе арабовой кислоты получаются L-арабиноза (34,4%), D-галактоза (29,5%), L-рамноза (14,2%) и альдобиуроновая кислота (28,3%), состоящая из галактозы и глюкуроновой кислоты. Важные данные о строении арабовой кислоты были получены при ее ступенчатом гидролизе.
5.4. Нуклеозиды, нуклеотиды, нуклеиновые кислоты
Нуклеоти́ды — фосфорные эфиры нуклеозидов, нуклеозидфосфаты. Свободные нуклеотиды, в частности АТФ, цАМФ, АДФ, играют важную роль в энергетических и информационных внутриклеточных процессах, а также являются составляющими частями нуклеиновых кислот и многих коферментов.
Нуклеотиды являются сложными эфирами нуклеозидов и фосфорных кислот. Нуклеозиды, в свою очередь, являются N-гликозидами, содержащими гетероциклический фрагмент, связанный через атом азота с C-1 атомом остатка сахара.
Строение нуклеотидов
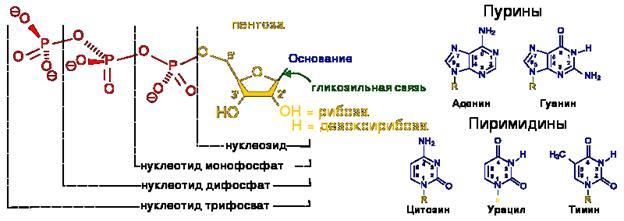
В природе наиболее распространены нуклеотиды, являющиеся β-N-гликозидами пуринов или пиримидинов и пентоз - D-рибозы или D-2-рибозы. В зависимости от структуры пентозы различают рибонуклеотиды и дезоксирибонуклеотиды, которые являются мономерами молекул сложных биологических полимеров (полинуклеотидов) — соответственно РНК или ДНК.[1]
Фосфатный остаток в нуклеотидах обычно образует сложноэфирную связь с 2'-, 3'- или 5'-гидроксильными группами рибонуклеозидов, в случае 2'-дезоксинуклеозидов этерифицируются 3'- или 5'-гидроксильные группы.
Большинство нуклеотидов являются моноэфирами ортофосфорной кислоты, однако известны и диэфиры нуклеотидов, в которых этерифицированы два гидроксильных остатка - например, циклические нуклеотиды циклоаденин- и циклогуанин монофосфаты (цАМФ и цГМФ). Наряду с нуклеотидами - эфирами ортофосфорной кислоты (монофосфатами) в природе также распространены и моно- и диэфиры пирофосфорной кислоты (дифосфаты, например, аденозиндифосфат) и моноэфиры триполифосфорной кислоты (трифосфаты, например, аденозинтрифосфат).
Соединения, состоящие из двух нуклеотидовых молекул, называются динуклеотидами, из трёх — тринуклеотидами, из небольшого числа — олигонуклеотидами, а из многих — полинуклеотидами, или нуклеиновыми кислотами.
Названия нуклеотидов представляют собой аббревиатуры в виде стандартных трёх- или четырёхбуквенных кодов.
Если аббревиатура начинается со строчной буквы «д» (англ. d), значит подразумевается дезоксирибонуклеотид; отсутствие буквы «д» означает рибонуклеотид. Если аббревиатура начинается со строчной буквы «ц» (англ. c), значит речь идёт о циклической форме нуклеотида (например, цАМФ).
Первая прописная буква аббревиатуры указывает на конкретное азотистое основание или группу возможных нуклеиновых оснований, вторая буква — на количество остатков фосфорной кислоты в структуре (М — моно-, Д — ди-, Т — три-), а третья прописная буква — всегда буква Ф («-фосфат»; англ. P).
Латинские и русские коды для нуклеиновых оснований:
A — А: Аденин;
G — Г: Гуанин;
C — Ц: Цитозин;
T — Т: Тимин (5-метилурацил), не встречается в РНК, занимает место урацила в ДНК;
U — У: Урацил, не встречается в ДНК, занимает место тимина в РНК.
Общепринятые буквенные коды для обозначения нуклеотидных оснований соответствуют номенклатуре, принятой Международным союзом теоретической и прикладной химии (International Union of Pure and Applied Chemistry, сокращённо — англ. IUPAC, русск. ИЮПАК) и Международным союзом биохимии и молекулярной биологии (International Union of Biochemistry and Molecular Biology, сокращённо — англ. IUBMB). Если при секвенировании последовательности ДНК или РНК возникает сомнение в точности определения того или иного нуклеотида, помимо пяти основных (A, C, T, G, U), используют другие буквы латинского алфавита в зависимости от того, какие наиболее вероятные нуклеотиды могут находиться в данной позиции последовательности.
Длину секвенированных участков ДНК (гена, сайта, хромосомы) или всего генома указывают в парах нуклеотидов (пн), или парах оснований (англ. base pairs, сокращённо bp), подразумевая под этим элементарную единицу двухцепочечной молекулы нуклеиновой кислоты, сложенную из двух спаренных комплементарных оснований.
Нуклеи́новые кисло́ты (от лат. nucleus — ядро) — высокомолекулярные органические соединения, биополимеры (полинуклеотиды), образованные остатками нуклеотидов. Нуклеиновые кислоты ДНК и РНК присутствуют в клетках всех живых организмов и выполняют важнейшие функции по хранению, передаче и реализации наследственной информации.
Химические свойства
Нуклеиновые кислоты хорошо растворимы в воде, практически не растворимы в органических растворителях. Очень чувствительны к действию температуры и критических значений уровня pH. Молекулы ДНК с высокой молекулярной массой, выделенные из природных источников, способны фрагментироваться под действием механических сил, например при перемешивании раствора. Нуклеиновые кислоты фрагментируются ферментами — нуклеазами.
Строение
Фрагмент полимерной цепочки ДНК
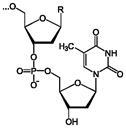
Полимерные формы нуклеиновых кислот называют полинуклеотидами. Цепочки из нуклеотидов соединяются через остаток фосфорной кислоты (фосфодиэфирная связь). Поскольку в нуклеотидах существует только два типа гетероциклических молекул, рибоза и дезоксирибоза, то и имеется лишь два вида нуклеиновых кислот — дезоксирибонуклеиновая (ДНК) и рибонуклеиновая (РНК).
Мономерные формы также встречаются в клетках и играют важную роль в процессах передачи сигналов или запасании энергии. Наиболее известный мономер РНК — АТФ, аденозинтрифосфорная кислота, важнейший аккумулятор энергии в клетке.
ДНК — Дезоксирибонуклеиновая кислота. Сахар — дезоксирибоза, азотистые основания: пуриновые — гуанин (G), аденин (A), пиримидиновые — тимин (T) и цитозин (C). ДНК часто состоит из двух полинуклеотидных цепей, направленных антипараллельно.
РНК — Рибонуклеиновая кислота. Сахар — рибоза, азотистые основания: пуриновые — гуанин (G), аденин (A), пиримидиновые урацил (U) и цитозин (C). Структура полинуклеотидной цепочки аналогична таковой в ДНК. Из-за особенностей рибозы молекулы РНК часто имеют различнные вторичные и третичные структуры, образуя комплементарные участки между разными цепями.
5.5 Липиды
Липи́ды (от греч. λίπος, lípos — жир) — жирные кислоты, а также их производные, как по радикалу, так и по карбоксильной группе.
Используемое ранее определение липидов, как группы органических соединений, хорошо растворимых в неполярных органических растворителях (бензол, ацетон, хлороформ) и практически нерастворимых в воде, является неточным. Во-первых, такое определение вместо четкой характеристики класса химических соединений говорит лишь о физических свойствах. Во-вторых, в настоящее время известно достаточное количество соединений, нерастворимых в неполярных растворителях или же, наоборот, хорошо растворимых в воде, которые, тем не менее, относят к липидам. В современной органической химии определение термина «липиды» основано на биосинтетическом родстве данных соединений — к липидам относят жирные кислоты и их производные [1]. В то же время в биохимии и других разделах биологии к липидам по-прежнему принято относить и гидрофобные или амфифильные вещества другой химической природы.
Молекулы простых липидов состоят из спирта, жирных кислот, сложных - из спирта, высокомолекулярных жирных кислот, возможны остатки фосфорной кислоты, углеводов, азотистых оснований и др. Строение липидов зависит в первую очередь от пути их биосинтеза.
6 Строение и основные химические свойства групп соединений растительного и животного происхождения
ТЕРПЕНЫ, группа преим. ненасыщ. углеводородов состава (C5H8)n, где n2; широко распространены в природе (гл. обр. в растит., реже в животных организмах). Все терпены обычно рассматривают как продукты полимеризации изопрена (см. Изопреноиды), хотя биосинтез их иной: протекает аналогично биосинтезу карбоковых к-т, т.е. через ацетилкоэнзим А и ацетоацетилкоэнзим А. Дальнейшие биохим. превращения приводят к образованию мевалоновой к-ты, к-рая в результате ферментативного фосфорилирования, декарбокси-лирования и дегидратаций переходит в изопентенилпирофосфат, изомеризующийся затем в диметилаллилпирофос-фат. Два последних, взаимодействуя друг с другом, образуют геранилпирофосфат, к-рый далее алкилирует изопенте-нилпирофосфат до фарнезилпирофосфата; эти С10- и С15-соед. являются ключевыми при биосинтезе всех терпенов (см. также Обмен веществ).
По числу изопреновых звеньев терпены подразделяют на: монотерпены, или собственно терпены С10Н16 (часто только эти в-ва подразумевают под терпенами, напр. лимонен, мирцен); сесквитер-пены, или полуторатерпены С15К24 (напр., бизаболен); ди-терпены и их производные С20Н32 (напр., смоляные кислоты-абиетиновая, левопимаровая и др.); тритерпены С30Н48 (напр., нек-рые гормоны и стерины-ланостерин, олеаяоловая к-та, сквален и т. д.); политерпены (см. Каучук натуральный).
Каждый ряд терпенов разделяется на группы:
1) алифатические, или ациклические,-соед. с открытой цепью углеродных атомов; монотерпены этой группы включают три двойные связи (напр., аллооцимен, оци-мен).
2) Карбоциклические - содержат одно или неск. колец углеродных атомов. По числу колец различают: а) моноциклические, собственно терпены данной группы включают две двойные связи (ментадиены, в т. ч. терпинены, терпинолен и др.); б) бициклические, монотерпены этой группы содержат только одну двойную связь (см. Камфен, Карены, Пинены); в) трициклические, монотерпены данной группы не содержат двойных связей (напр., трициклен); г) сесквитер-пены, дитерпены, тритерпены и политерпены могут содержать и более трех циклов.
Сопутствующие обычно терпенам их производные часто наз. терпеноидами, по характеру функц. групп они разделяются на спирты, альдегиды, кетоны, сложные эфиры, пероксиды, к-ты и т.д. [напр., борнеол, камфора, (-)-ментол, терпинеолы].
Монотерпены и сесквитерпены часто обладают довольно приятным запахом. Особенно нежный запах характерен для их кислородных производных (спирты, альдегиды, сложные эфиры); именно они вместе с терпенами обусловливают аромат цветов, запах хвойных и многих иных растений.
Терпены весьма реакционноспособны: легко окисляются на воздухе, особенно на свету, часто превращаясь при этом в кислородсодержащие соед.; при нагр. изомеризуются (прежде всего при взаимод. с кислыми агентами); диспропорциони-руют в присут. катализаторов (Pd, Pt, Ni); по двойным связям легко гидрируются, гидра тируются, присоединяют галогены, галогеноводороды, орг. к-ты и т. д. При сильном нагревании без доступа воздуха (400-500 °С) кольца терпенов раскрываются, причем из бициклических терпенов можно получить моноциклические и даже алифатические (см. Камфеновые перегруппировки). При нагр. до 700 °С и выше все терпены разлагаются с образованием сложной смеси продуктов (изопрен, ароматич. углеводороды и др.).
6.2 Стероиды
СТЕРОИДЫ, группа природных и синтетических химических соединений – производных частично или полностью гидрированного 1,2-циклопентенофенантрена типа

в молекулярном скелете которых 17 атомов углерода образуют 4 сочлененных кольца A, B, C, D. Стероиды широко распространены в природе, они участвуют в осуществлении самых разнообразных биологических функций. Стероидную природу имеют половые гормоны, витамин D, гормоны надпочечников, желчные кислоты, гормоны линьки и метаморфоза членистоногих, репелленты насекомых, отпугивающие хищников, и яды в коже жаб. И природные, и синтетические стероиды при сходном строении проявляют сильно различающееся физиологическое действие, поэтому они широко применяются в медицине в качестве противовоспалительных, сердечных, противозачаточных и других средств.
подразделяют на стерины, желчные кислоты, стероидные гормоны, стероидные сапонины, сердечные гликозиды и стероидные алкалоиды (см. также АЛКАЛОИДЫ).
Чаще всего стероиды встречаются в форме стеринов, обнаруженных практически во всех растениях, грибах и животных. Это, в частности, известный всем холестерин, который служит исходным веществом для синтеза в организме всех стероидных гормонов. Холестерин может использоваться для промышленного получения многих стероидов, однако экономически выгоднее применять вместо него некоторые из легкодоступных растительных стеринов (например, стигмастерин из соевых бобов), имеющих структурное сходство с целевыми стероидами.
6.3 Алкалоиды
Алкало́иды — группа азотсодержащих органических соединений природного происхождения (чаще всего растительного), большинство из которых обладает свойствами слабого основания. Некоторые нейтральныеи даже слабокислотные соединения также относятся к алкалоидам. Иногда алкалоидами называются и синтетические соединения аналогичного строения.
Помимо углерода, водорода и азота в молекулы алкалоидов могут входить атомы серы, реже — хлора, брома или фосфора. Многие алкалоиды обладают выраженной физиологической активностью. К алкалоидам относятся, например, такие вещества, как морфин, кофеин, кокаин, стрихнин, хинин и никотин.
Граница между алкалоидами и другими азотсодержащими природными соединениями различными авторами проводится по-разному. Такие соединения, как аминокислоты, пептиды, белки, нуклеотиды, нуклеиновые кислоты, аминосахара и антибиотики обычно не называются алкалоидами. Иногда считается, что природные соединения, содержащие азот в экзоциклической позиции (мескалин, серотонин, дофамин и др.), относятся к аминам, но не к алкалоидам. Другие же авторы, напротив, считают алкалоиды частным случаем аминов или причисляют биогенные амины к алкалоидам
Алкалоиды, молекулы которых содержат атомы кислорода (что справедливо для подавляющего большинства алкалоидов) при стандартных условиях, как правило, представляют собой бесцветные кристаллы. Алкалоиды, молекулы которых не содержат атомов кислорода, чаще всего являются летучими бесцветными маслянистыми жидкостями (как никотин или кониин). Некоторые алкалоиды не являются бесцветными: так, берберин жёлтый, сангвинарин оранжевый.
Большинство алкалоидов обладает свойствами слабых оснований, но некоторые из них амфотерны (как теобромин и теофиллин).
Как правило, алкалоиды плохо растворимы в воде, но хорошо растворимы во многих органических растворителях (диэтиловом эфире, хлороформе и 1,2-дихлорэтане). Исключением является, например, кофеин, хорошо растворимый в кипящей воде. При взаимодействии с кислотами алкалоиды образуют соли различной степени прочности. Соли алкалоидов, как правило, хорошо растворимы в воде и спиртах и плохо растворимы в большинстве органических растворителей, хотя известны соли, плохо растворимые в воде (сульфат хинина) и хорошо растворимые в органических растворителях (гидробромид скополамина).
6.4 Жиры и растительные масла
ЖИРЫ, в-ва животного (см. Жиры животные), растительного (см. Растительные масла) и микробного происхождения, состоящие в осн. (до 98%) из триглицеридов (ацилглицеринов) полных эфиров глицерина и жирных к-т. Содержат также ди- и моноглицериды (1-3%), фосфолипиды, гликолипиды и диольные липиды (0,5-3%), своб. жирные к-ты, стерины и их эфиры (0,05 1,7%), красящие в-ва (каротин, ксантофилл), витамины A, D, Е и К, полифенолы и их эфиры. Хим., физ. и биол. св-ва жиров определяются входящими в его состав триглицеридами и, в первую очередь, длиной цепи, степенью ненасыщенности жирных к-т и их расположением в триглицериде. В состав жиров входят в осн. неразветвленные жирные к-ты, содержащие четное число атомов С (от 4 до 26) как насыщенные, так моно- и полиненасыщенные; в осн. это миристиновая, пальмитиновая, стеариновая, 9-гексадеценовая, олеиновая, линолевая и линоленовая к-ты. Почти все ненасыщ. к-ты растит. жиров и большинства животных жиров являются цис-изомерами. Жиры жвачных животных содержат транс-изомеры. Триглицериды, содержащие остатки разл. к-т, существуют в виде неск. изомеров положения, а также в виде разл. стереоизомеров, напр.:

Триглицериды прир. жиров содержат по крайней мере две разл. жирные к-ты. Различают Триглицериды, содержащие три насыщ. к-ты (S3), две насыщ. и одну ненасыщ. (соотв. SSU и SUS), одну насыщ. и две ненасыщ. (соотв. SUU и USU) и три ненасыщ. к-ты (U3) (см. табл.).
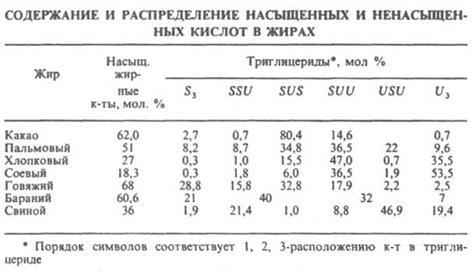
Химические свойства. Гидролиз жиров, конечные продукты к-рого глицерин и жирные к-ты, осуществляют в пром-сти нагреванием их с водой до 200-225 °С при 2-2,5.106 Па (безреактивный способ) или нагреванием при нормальном давлении в присут. сульфокислот (катализатор Твитчела и контакт Петрова). Щелочной катализ применяют в процессах мыловарения (см. Мыла) и при наличии в жирнокислотных цепях гидроксильных групп. Скорости ферментативного гидролиза a- и b-сложноэфирных групп ферментом панкреатич. липазой различны, что используют для установления строения триглицеридов жиров. Алкоголиз жиров, в частности метанолиз, используется как первая ступень непрерывного метода мыловарения. Глицеролиз действием глицерина применяют для получения моно-и диглицеридов, используемых в качестве эмульгаторов. Ацидолиз, напр., ацетолиз кокосового жира с послед. этерификацией избытка уксусной к-ты глицерином, приводит к смеси, состоящей из лауроилдиацетина, миристоилдиацетина и др. смешанных триглицеридов, применяемой в качестве мягчителей нитроцеллюлозы. Большое практич. значение имеет р-ция двойного обмена ацильными радикалами в триглицеридах (переэтерификация), протекающая как внутри-, так и межмолекулярно и приводящая к перераспределению остатков жирных к-т. При проведении этой р-ции в однофазной жидкой системе (ненаправленная переэтерификация) происходит статистич. перераспределение кислотных остатков в образующейся смеси триглицеридов. Направленная (многофазная) переэтерификация осуществляется при такой т-ре, при к-рой высокоплавкие триглицериды находятся в твердом, а низкоплавкие - в жидком состоянии. При направленной переэтерификации жиры обогащаются наиб. высокоплавкими (S3) и наиб. низкоплавкими (U3) триглицеридами. Ненаправленная и особенно направленная переэтерификация натуральных жиров используется для изменения их физ. св-в - т-ры плавления, пластичности, вязкости. Ацидолиз и алкоголиз жиров проводят преим. в присут. кислотных катализаторов, переэтерификацию - в присут. основных. Большое значение имеют восстановление (см. Гидрогенизация жиров) и цис-, транс-изомеризация непредельных ацильных остатков триглицеридов. Изомеризацию цис-изомеров ненасыщ. к-т в транс-изомеры (э л а и д и р о в а н и е) проводят при 100-200 °С в присут. кат. - Ni, Se, оксидов N, S. При изомеризации полиненасыщ. к-т (рыбий жир) образуются к-ты с сопряженными двойными связями, обладающие высокой способностью к высыханию. Прогоркание жиров, проявляющееся в появлении специфич. запаха и неприятного вкуса, вызвано образованием низкомол. карбонильных соед. и обусловлено рядом хим. процессов. Различают два вида прогоркания - биохим. и химическое. Биохим. прогоркание характерно для жиров, содержащих значительное кол-во воды и примеси белков и углеводов (напр., для коровьего масла). Под воздействием содержащихся в белках ферментов (липаз) происходит гидролиз жиров и образование своб. жирных к-т. Увеличение кислотности может не сопровождаться появлением прогорклости. Микроорганизмы, развивающиеся в жирах, выделяют др. ферменты - липооксидазы, под действием к-рых жирные к-ты окисляются до b-кетокислот. Метилалкилкетоны, образующиеся при распаде последних, являются причиной изменения вкуса и запаха жиров. Во избежание этого производится тщательная очистка жиров от примесей белковых в-в, хранение в условиях, исключающих попадание микроорганизмов, и при низкой т-ре, а также добавка консервантов (NaCl, бензойная к-та). Хим. прогоркание - результат окисления жиров под действием О2
воздуха (автоокисление). Первая стадия - образование пероксильных радикалов при атаке молекулярным О2
углеводородных остатков как насыщ., так и ненасыщ. жирных к-т. Р-ция промотирустся светом, теплом и соед., образующими своб. радикалы (пероксиды, переходные металлы). Пероксильные радикалы инициируют неразветвленные и разветвленные цепные р-ции, а также распадаются с образованием ряда вторичных продуктов - гидроксикислот, эпоксидов, кетонов и альдегидов. Последние и вызывают изменение вкуса и запаха жиров. Для жиров, в к-рых преобладают насыщ. жирные к-ты, характерно образование кетонов (кетонное прогоркание), для жиров с высоким содержанием ненасыщ. к-т - альдегидное прогоркание. Для замедления и предотвращения хим. прогоркания используют ингибиторы радикальных р-ций: смесь 2- и 3-трет-бутил-4-гидроксианизола (БОА), 3,5-ди-трет-бутил-4-гидрокситолуол (БОТ), эфиры галловой к-ты, а также соед., образующие комплексы с тяжелыми металлами (напр., лимонная, аскорбиновая к-ты).
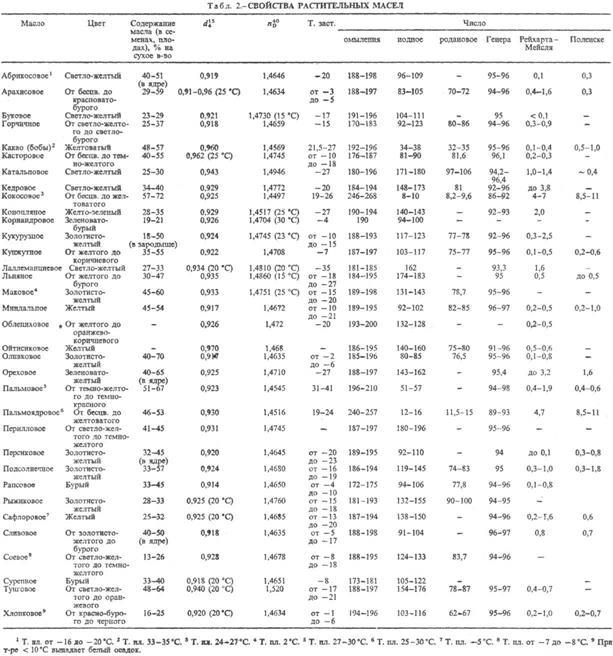
РАСТИТЕЛЬНЫЕ МАСЛА жирные (жиры растительные), продукты, извлекаемые из растит. сырья и состоящие в осн. из триглицеридов высших жирных к-т. Осн. источники растительных масел - масличные растения (масличные культуры). Растительные масла содержатся также в косточках нек-рых плодовых деревьев (абрикос, персик, вишня, черешня, миндаль), семенах винограда, арбуза, томатов, табака, чая, а также в разл. маслосодсржащих отходах пищ. произ-в, перерабатывающих с.-х. сырье. К последним относят гл. обр. отруби и зародыши семян зерновых культур. В оболочке зерна пшеницы и ржи содержится 5-6% масла, в зародыше-11-13 и 10-17% соотв.; в зародыше кукурузы 30-48% масла, проса-ок. 27%, риса-24-25%. Содержание масла в растениях и его качество зависят от сорта растения, условий произрастания (удобрения, обработка почвы), степени зрелости плодов и семян.
Свойства. Растительные масла на 94-96% состоят из смесей триглицеридов высших жирных кислот (табл. 1). Оставшуюся часть составляют в-ва, близкие к жирам (напр., фосфо-липиды, стерины, витамины), своб. жирные к-ты и др. компоненты.
Плотность растительных масел 0,87-0,98 г/см3 (табл. 2); большинство из них раств. в бензине, бензоле, дихлорэтане, сероуглероде, ацетоне, диэтиловом эфире, ССl4
; ограниченно раств. в этаноле и метаноле, не раств. в воде.
Св-ва растительных масел определяются гл. обр. составом и содержанием жирных к-т, образующих триглицериды (см. Жиры). Обычно это насыщ. и ненасыщ. одноосновные жирные к-ты с неразветвленной углеродной цепью и четным числом атомов углерода (преим. С16
и С18
). В подавляющем большинстве растительные масла содержат смеси глицеридов разл. к-т, в нек-рых присутствуют и глицериды одной к-ты. Кроме того, в растительных маслах обнаружены в небольших кол-вах глицериды жирных к-т с нечетным числом атомов углерода.
В зависимости от состава триглицеридов растительные масла могут быть жидкими (подсолнечное, хлопковое, соевое, рапсовое, кукурузное, льняное и др.) и твердыми (кокосовое, пальмовое, пальмоядровое и др.). У жидких масел, содержащих гл. обр. непредельные к-ты, т-ра застывания ниже 0°С, у твердых - достигает 40 °С. При контакте с О2
воздуха или при нагр. до 250-300°С многие растительные масла подвергаются окислит. полимеризации ("высыхают"), образуя пленки. По способности к высыханию растительные масла условно подразделяют на высыхающие, полувысыхающие и невысыхающие. Первые, напр. льняное масло, конопляное и тунговое масла, содержат гл. обр. триглицериды к-т с двумя или тремя двойными связями (линолевой, линоленовой, элеостеариновой); вторые, напр. подсолнечное масло, соевое и маковое масла,-триглицериды к-т с одной или двумя двойными связями (олеиновой, линолевой); третьи, напр. кокосовое и пальмовое масла,-преим. триглицериды насыщ. к-т (лауриновой, пальмитиновой, стеариновой) и небольшое кол-во монрненасыщ. олеиновой. Невысыхающее касторовое масло содержит тригли-церид рицинолевой к-ты.
При анализе состава растительных масел кол-во высших жирных к-т, образующихся при омылении, характеризуют числом омыления, степень ненасыщенности - йодным и родановым числами.
Компоненты растительных масел, отличные от триглицеридов, подразделяют на омыляемые и неомыляемые. К первым относят своб. жирные к-ты (содержание 1-2%), фосфолипиды (0,5-4%), стерины (0,3-1,3%), воски и воскообразные в-ва (0,002-0,4%), пигменты (не более 0,16%), ко вторым-белки (0,1-1,5%), витамины (до 0,5%), углеводороды и др.
Свободные жирные к-ты могут содержаться в растит. сырье (семена недозревших растений или семена, самосозревающие при хранении во влажном состоянии) или образовываться в процессе выделения масла в результате частичного гидролиза триглицеридов (высшие жирные к-ты) и их окисления под действием света и при длит. хранении (низкомол. жирные к-ты - масляная, каприновая, капроновая, каприло-вая, ацетоуксусная, уксусная). Суммарное содержание своб. к-т в % по массе в растительных маслах определяет их кислотность и характеризуется кислотным числом. Наличие своб. низкомол. жирных к-т, р-римых в воде и испаряющихся при нагр., характеризуется числом Рейхарта-Мейсля; наличие к-т, не растворяющихся в воде, но способных испаряться при нагр.,-числом Поленске. Оба этих числа определяются кол-вом мл 0,1 н. р-ра КОН, расходуемого на нейтрализацию 5 г растительного масла в определенных условиях. Содержание нерастворимых к-т и неомыляемых компонентов характеризуется числом Генера (содержание их в % в 100 г растительного масла).

Фосфолипиды в растительных маслах представлены гл. обр. глицеро-фосфатидами (лецитины), в меньшем кол-ве -инозитфосфа-тидами и сфингомиелинами. Фосфолипиды растительных масел участвуют в биол. окислении масел в организме и сами по себе представляют большую ценность (см. Фосфолипиды). Однако в растительных маслах они образуют коллоидные р-ры, из к-рых при поглощении воды коагулируют с образованием осадков, наз. фузами. В таких осадках могут происходить гидроли-тич. процессы, приводящие к потере масел и затруднениям при переработке. Под действием О2 воздуха фосфолипиды легко окисляются с образованием темноокрашенных соед., ухудшающих качество масел. Поэтому растительные масла, не идущие непосредственно в пищу или подвергающиеся дальнейшей переработке (напр., рафинированию), очищают-вт фосфоли-пидов, подвергая масло гидратации, или связывая с помощью разл. хим. агентов, напр. диметилдиаллиламмоний-хлорида. Выделенные фосфолипиды, учитывая их биол. и пищ. ценность, используют для произ-ва фосфолипидных концентратов, к-рые добавляют во мн. пищ. продукты (напр., маргарин) и корма для животных.
Из стеринов растит. происхождения (фитостеринов) в растительных маслах наиб. часто содержатся ситостерин и стигмастерин, являющиеся предшественниками витамина D (см. Стерины). Холестерин в растительных маслах практически не содержится. Наиб. кол-во стеринов содержится в кукурузном масле-0,42-1,38%, в подсолнечном их 0,25-0,53%, в хлопковом 0,26-0,57%, в соевом 0,35-0,40%. При переработке и очистке растительных масел потери стеринов стараются свести к минимуму. При необходимости стерины из растительных масел могут быть извлечены с помощью алкалоида дигитонина, с к-рым они дают нерастворимые в этаноле соединения.
Воски и воскообразные вещества в растительных маслах образуют эмульсии и вызывают помутнение масла. Для их удаления масло обычно охлаждают до 8-12°С и осадок отфильтровывают (способ "вымораживания").
Пигменты, содержащиеся в семенах и плодах масличных растений, придают растительным маслам разл. окраску. Красные и желтые оттенки в цвете растительных масел определяются присутствием в них каротиноидов (красный оттенок-каротин, желтый-ксантофилл), наиб. их кол-во содержится в кукурузном масле (0,058-0,15%). Зеленый оттенок, характерный для соевого, кукурузного, рапсового, горчичного и др. масел, определяется присутствием в них смеси хлорофиллов А и В. В хлопковом масле содержится токсичный пигмент госсипол (0,14-2,5% по массе), наиб. содержание к-рого отмечается в масле, подученном из низкосортных и незрелых хлопковых семян. При переработке масла госсипол дает разл. темно-окрашенные продукты. Удаляют госсипол из масла с помощью антраниловой к-ты, с к-рой он образует нерастворимое соединение. При очистке растительных масел с помощью адсорбентов происходит удаление пигментов и осветление масла.
Осн. массу белковых веществ, переходящих в растительные масла из семян, составляют альбумины и глобулины. Поскольку наличие белков ухудшает товарный вид масел и увеличивает его потери при очистке и хранении, белковые примеси (вместе с фосфолипидами) удаляют при гидратации масла, а также под действием щелочей или минер. к-т. Углеводы, моно-, ди- и олигосахариды, декстрины, крахмал, клетчатка и гемицеллюлоза, содержащиеся в растительных маслах в кол-ве 0,02-0,5%, образуют стабильные эмульсии, способствуют потемнению масла при термич. обработке, придают маслам неприятный вкус и запах.
Часть неомыляемых в-в, входящих в растительные масла, составляют витамины Е, A, D и К. Витамин Е содержится в растительных маслах в виде a-, b-, g-, и d-токоферолов. Кол-во D-a-токоферола в подсолнечном масле составляет ок. 0,05%. Высоким содержанием токоферолов характеризуются также масла пшеничных отрубей (100-400 мг в 100 г масла), соевое (74-160 мг в 100 г масла) и кукурузное (87-200 мг) масла; до 100 мг токоферолов в 100 г подсолнечного, хлопкового, рапсового и нек-рых др. маслах, до 60 мг-в арахисовом, до 30 мг-в оливковом и кокосовом.
Витамин А встречается в растительных маслах в виде провитаминов; содержится преим. в облепиховом, абрикосовом, персиковом и др. маслах. Витамин D содержится гл. обр. в соевом и кунжутном маслах, витамин К (К1, К2, К3)-в конопляном, подсолнечном, льняном и сурепном маслах.
В растительных маслах присутствуют также незначит. кол-ва насыщ. и не-насыщ. углеводородов с разветвленной цепью. В частности, в состав подсолнечного, хлопкового и соевого масел входит сквален (0,008-0,012%). Углеводороды, совместно с белками, в значит. степени определяют вкус и запах масла.
В результате длит. хранения на свету, при повыш. т-ре или под действием микроорганизмов растительные масла портятся-прогоркают. Неприятный запах и вкус растительным маслам сообщают продукты окисления жирных к-т (альдегиды, кетоны, гидроксикисло-ты), низкомол. жирные к-ты и их глицериды, продукты распада каротиноидов, стеринов, витаминов, фосфолипи-дов.
Иногда в растительных маслах могут находиться пестициды, используемые в с. х-ве. Их обычно удаляют из масла вместе с одорирующими в-вами в процессе перегонки с паром при 200-250 °С в вакууме.
6.5 Воски. Спермацет
ВОСКИ, исторически сложившееся название разных по составу и происхождению продуктов, преим. природных, к-рые по св-вам близки пчелиному воску. Природные воски Представляют собой пластичные легко размягчающиеся при нагр. продукты, большинство из к-рых плавится в интервале 40-90°С (см. табл.). Нек-рые воски, напр. пчелиный и буроугольный, являются гетерогенными системами, в к-рых дисперсная кристаллич. фаза распределена в аморфной дисперсионной среде. Воски не смачиваются водой, водонепроницаемы, обладают низкой электрич. проводимостью, горючи. Они не раств. в холодном этаноле, хорошо раств. в бензине, хлороформе, бензоле и диэтиловом эфире. Большинство прир. восков содержит сложные эфиры одноосновных насыщенных карбоновых к-т нормального строения и спиртов с 12-46 атомами С в молекуле. Такие воски по хим. св-вам близки к жирам (триглицеридам), но омыляются только в щелочной среде. Иногда прир. продукты, не содержащие сложные эфиры, напр. парафин, петролатум, церезин, наз. аналогами восков или воскоподобными материалами.
воск-смесь сложных эфиров (72%), насыщенных неразветвленных углеводородов С21
—С35
(12-15%) и карбоновых к-т С16
—С36
(15%), относит. кол-ва к-рых зависят от условий питания пчел и др. факторов. Получают переработкой сот, обрезков вощины и восковых наростов в ульях.
Шерстяной (шерстный) воск выделяется кожными железами овец в волосяную луковицу и обильно покрывает шерсть (в кол-ве 5-16% по массе). В его состав входят: сложные эфиры жирных к-т и высших спиртов, в т. ч. ланолинового С11
Н21
СН2
ОН; жирные к-ты (12-40%); спирты (44-45%); углеводороды (14-18%); стерины (холестерин, изохолестерин, эргостерин) в своб. виде и в виде сложных эфиров (10%). Получают из промывных вод шерстомоек или экстрагированием шерсти орг. р-рителями. После обработки щелочами, отбелки окислителями и адсорбентами получают очищенный шерстяной воск-ланолин. Последний в отличие от др. восков образует устойчивые эмульсии с водой, взятой в кол-ве, превышающем массу воска в 1,8-2 раза.
цет содержится вместе со спермацетовым маслом в костных черепных углублениях нек-рых видов китов, особенно кашалотов. Состоит на 98% из цетина С15
Н31
СООС16
Н33
. Спермацет отделяют от масла вымораживанием. Гидрируя спермацетовое масло, получают воск, близкий по св-вам спермацету.
Китайский воск вырабатывается червецом Coccus ceriferus, к-рый обитает гл. обр. на китайском ясене и образует на нем восковой покров. Содержит сложный эфир гексакозановой к-ты СН3
(СН2
)24
СООН и гексадеканового спирта СН3
(СН2
)15
ОН (95-97%), смолу (до 1%), углеводороды (до 1%) и спирты (до 1%).
Шеллачный воск содержится в прир. смоле - шеллаке (ок. 5%). В него входят 60-62% сложных эфиров, 33-35% спиртов, 2-6% углеводородов. Выделяют при охлаждении спиртового р-ра шеллака.
Воск бактерий покрывает пов-сть кислотоупорных бактерий, напр. туберкулезных и лепры, обеспечивая их устойчивость к внеш. воздействиям. Содержит сложные эфиры миколевой к-ты С88
Н172
О4
иэйкозанола СН3
(СН2
)17
СНОНСН3
, а также октадеканола СН3
(СН2
)15
СНОНСН3
.
Воск сахарного тростника покрывает тонкой пленкой стебли растений. В него входят сложные эфиры (78-82%), насыщенные С14
—С34
и ненасыщенные С15
—С37
углеводороды (3-5%), насыщенные жирные к-ты С12
—С36
(14%) и спирты С24
—С34
(6-7%). При отжиме тростника ок. 60% воска переходит в сок. При очистке последнего воск выпадает в осадок, из к-рого его извлекают экстракцией орг. р-рителями.
СВОЙСТВА ВОСКОВ
Карнаубский воск покрывает листья пальмы Copernicia cerifera. Состоит на 80% из сложных эфиров триаконтанола CH3
(CH2
)29
OH и тетракозановой СН3
(СН2
)22
СООН и гексакозановой к-т. Содержит также 10% спиртов - октакозанола СН3
(СН2
)27
ОН, гептакозанола СН3
(СН2
)26
ОН, не встречающегося в остальных восках, и др., а также 1-1,5% углеводородов, 0,5% фитостерина. Для получения воска листья пальмы высушивают, из них выколачивают порошок, к-рый вываривают в воде и выливают в формы. 2000 листьев дают ок. 16 кг воска.
Пальмовый воск находится в углублениях кольчатого ствола восковой пальмы Ceroxilon ondlicoka, откуда его соскабливают. Состоит преим. из сложных эфиров гексакоза-новой к-ты с гексакозанолом СН3
(СН2
)25
ОН и триаконта-нолом СН3
(СН2
)29
ОН. Одно дерево дает ок. 12кг воска.
Канделильский воск извлекают из травы канделилы Pedilanthus Pavonis Boas, растущей в Мексике. С 1 га получают от 2 до 8 т воска, к-рый содержит до 40% углеводородов. Японский воск добывают из лакового дерева Rhus vernicifera, произрастающего в Японии и Китае. Содержит глицериды гексадекановой, октадекановой, эйкозановой СН3
(СН2
)18
СООН и нек-рых дикарбоновых к-т, а также карбоновые к-ты и спирты. Получают вывариванием в воде мучнистой массы, образующейся при измельчении косточек плодов.
Торфяной воск получают экстракцией бензином при 80 °С верхового битуминозного торфа со степенью разложения не менее 30%, влажностью не более 50% и зольностью не более 8% с послед. отдувкой р-рителя. Полученный продукт содержит 60-75% воска и 25-40% смол; состоит из сложных эфиров (50-52%), карбоновых к-т (35-40%), углеводородов (5-7%) и спиртов (2-3%). Смолы из воска экстрагируют бензином, охлажденным до 0-5 °С. Нерастворимую часть промывают р-рителем, продувают острым паром и получают обессмоленный воск. Различными методами очистки обессмоленного воскв получают рафинированный воск. Такой воск имеет кислотное число 160 и состоит на 97,5% из к-т С8—С30. Этерификацией его спиртами получают разл. виды этерифицированных восков.
Буроугольный воск (монтан-воск) экстрагируют бензолом или бензином из бурого битуминозного угля. Удалением смолы путем ее экстракции р-рителем получают обессмоленный воск, окислением последнего - рафинированный, этерификацией рафинированного воска одно-, двух-и многоатомными спиртами-этерифицированный воск. По составу буроугольный воск близок торфяному и отличается от него меньшим содержанием низкомол. кислородсодержащих соединений.
Озокерит (горный, или минеральный, воск)-минерал из группы нефтяных битумов; генетически связан с месторождениями парафинистой нефти. По хим. составу - смесь твердых (49,5%) и жидких (45%) насыщенных углеводородов и смол (5,5%). Экстрагируют из руды тяжелым бензином (т. кип. 100-200°С); оставшийся после отгонки р-рителя продукт фильтруют и отгоняют от него при 300°С легкие фракции. Обработкой озокерита 95-98%-ной H2
SO4
при 200°С под давлением с послед. нейтрализацией известью и очисткой отбеливающей глиной получают церезин. Пром-сть СССР выпускает церезины марок 80, 75, 69 и 57 (цифры указывают т-ру каплепадения), к-рые представляют собой смесь насыщенных углеводородов С37
Н76
-С53
Н108
гл. обр. изостроения.
Синтетические воски. В зависимости от типа исходного сырья делят на частично и полностью синтетические.
Воски частично синтетические получают окислением сырого монтан-воска смесью хромовой и серной к-т с послед. этерификацией продуктов окисления (восковых к-т) разл. гликолями. В эту группу также входят абрильские воски (смесь производных жирных к-т и алифатич. или ароматич. аминов) и воски на основе нефтяных и смоляных парафинов и их производных.
Воски полностью синтетические получают по р-ции Фишера-Тропша действием Н2
на СО. Образующиеся продукты состоят гл. обр. из высших алканов. Широкое применение находят также воски, состоящие из смеси полиолефинов (алкатены, виннотены, луполены) с мол. м. 2000-10000, степенью кристалличности 10-85%, плотн. 0,9-0,94 г/см3, вязкостью расплава при 140°С 0,085-1 кПа*с. В зависимости от мол. массы и кристалличности эти воски м. б. жидкими или твердыми.
Воски применяют более чем в 200 отраслях народного хозяйства. Они входят в состав политур, защитных композиций для металлов, тканей, бумаги, кож, дерева; применяются в литейной пром-сти как компоненты составов для изготовления форм при литье по выплавляемым моделям, смазок форм при получении изделий из пенополиуретанов, как изолирующий материал, компоненты мазей в косметике и медицине и др.
7 Строение, физические и химические свойства высокомолекулярных полимеризационных и поликонденсационных полимеров, используемых в фармации
Политетрафторэтиле́н, тефло́н или фторопла́ст-4 (-C2
F4
-)n
— полимер тетрафторэтилена (ПТФЭ), пластмасса, обладающая редкими физическими и химическими свойствами и широко применяемая в технике и в быту.
Слово «Тефлон®» является зарегистрированной торговой маркой корпорации DuPont. Непатентованное название вещества — «политетрафторэтилен» или «фторополимер».
Политетрафторэтилен был открыт в апреле 1938 года 27-летним учёным-химиком Роем Планкеттом [1], который случайно обнаружил, что закачанный им в баллоны под давлением газообразный тетрафторэтилен спонтанно полимеризовался в белый парафиноподобный порошок. Патент на изобретение тефлона принадлежит американской компании DuPont.
Свойства
Тефлон — белое, в тонком слое прозрачное вещество, по виду напоминающее парафин или полиэтилен. Обладает высокой тепло- и морозостойкостью, остается гибким и эластичным при температурах от —70 до +270 °C, прекрасный изоляционный материал. Тефлон обладает очень низкими поверхностным натяжением и адгезией и не смачивается ни водой, ни жирами, ни большинством органических растворителей.
По своей химической стойкости превышает все известные синтетические материалы и благородные металлы. Не разрушается под влиянием щелочей, кислот и даже смеси азотной и соляной кислот. Разрушается расплавами щелочных металлов, фтором и трифторидом хлора.
Тефлон применяют в химической, электротехнической и пищевой промышленности, в медицине, в военных целях, в основном, в качестве покрытий.
Полистирол — продукт полимеризации стирола (винилбензола) относится к полимерам класса термопластов.
Имеет химическую формулу вида: [-СН2
-С(С6
Н5
)Н-]n
-
Фенольные группы препятствуют упорядоченному расположению макромолекул и формированию кристаллических образований. Это жёсткий, хрупкий, аморфный полимер с высокой степенью оптического светопропускания, невысокой механической прочностью, выпускается в виде прозрачных гранул цилиндрической формы. Полистирол имеет низкую плотность (1060 кг/м3
), термическую стойкость (до 105 °С), усадка при литьевой переработке 0,4-0,8%. Полистирол обладает отличными диэлектрическими свойствами и неплохой морозостойкостью (до -40°C). Имеет невысокую химическую стойкость (кроме разбавленных кислот, спиртов и щелочей). Для улучшения свойств полистирола его модифицируют путём смешения с различными полимерами - подвергают сшиванию, таким образом получая сополимеры стирола.
Широкое применение полистирола (ПС) и пластиков на его основе базируется на его невысокой стоимости, простоте переработки и огромном ассортименте различных марок. Наиболее широкое применение (более 60% производства полистирольных пластиков) получили ударопрочные полистиролы, представляющие собой сополимеры стирола с бутадиеновым и бутадиен-стирольным каучуком. В настоящее время созданы и другие многочисленные модификации сополимеров стирола.
Растворяется в ацетоне, медленнее в бензине. Не растворим в воде. Термопластичный материал. Полистирол легко формуется и окрашивается. Хорошо обрабатывается механическими способами. Хорошо склеивается. Обладает низким влагопоглощением, высокой влагостойкостью и морозостойкостью.
Основные методы переработки: экструзия, литьё под давлением. Диапазон температур переработки лежит в пределах 190-240 °С. Из полистиролов производят широчайшую гамму изделий, которые в первую очередь применяются в бытовой сфере деятельности человека (одноразовая посуда, упаковка, детские игрушки и т. д.), а также строительной индустрии (теплоизоляционные плиты, несъемная опалубка, сандвич панели), облицовочные и декоративные материалы (потолочный багет, потолочная декоративная плитка, полистирольные звукопоглощающие элементы, клеевые основы, полимерные концентраты), медицинское направление (части систем переливания крови, чашки Петри, вспомогательные одноразовые инструменты). Вспенивающийся полистирол после высокотемпературной температурной обработки водой или паром может использоваться в качестве фильтрующего материала (фильтрующей насадки) в колонных фильтрах при водоподготовке и очистке сточных вод. Высокие электротехнические показатели полистирола в области сверхвысоких частот позволяют применять его в производстве: диэлектрических антенн, опор коаксиальных кабелей. Могут быть получены тонкие пленки (до 100 мкм), а в смеси с со-полимерами (стирол-бутадиен-стирол) до 20 мкм, которые также успешно применяются в упаковочной и кондитерской индустрии, а также производстве конденсаторов.
— термопластичный полимер этилена. Самый распространенный в мире пластик[1]. Представляет собой воскообразную массу белого цвета (тонкие листы прозрачны и бесцветны). Химически- и морозостоек, изолятор, не чувствителен к удару (амортизатор), при нагревании gjvjxcj.tjfnvxчрезвычайно низкая. Иногда в народном сознании отождествляется с целлофаном — похожим материалом растительного происхождения.
Полиэтилен высокого давления (ПЭВД), или Полиэтилен низкой плотности (ПЭНП) образуется при следующих условиях: температура 200-260°C; давление 150-300 МПа; присутствие инициатора (кислород или органический пероксид); в автоклавном или трубчатом реакторах. Реакция идёт по радикальному механизму. Получаемый по этому методу полиэтилен имеет средневесовой молекулярный вес 80 000—500 000 и степень кристалличности 50-60 %. Жидкий продукт впоследствии гранулируют. Реакция идёт в расплаве.
Полиэтилен среднего давления (ПЭСД) образуется при следующих условиях:
температура 100-120 °C;
давление 3—4 МПа;
присутствие катализатора (катализаторы Циглера—Натта (англ.), например, смесь TiCl4 и AlR3);
продукт выпадает из раствора в виде хлопьев. Получаемый по этому методу полиэтилен имеет средневесовой молекулярный вес 300 000—400 000, степень кристалличности 80-90 %.
Полиэтилен низкого давления (ПЭНД) или Полиэтилен высокой плотности (ПЭВП) образуется при следующих условиях:
температура 70-120 °C;
давление ниже 0.1 - 2 МПа;
присутствие катализатора (катализаторы Циглера—Натта, например, смесь TiCl4 и AlR3);
Полимеризация идёт в суспензии по ионно-координационному механизму. Получаемый по этому методу полиэтилен имеет средневесовой молекулярный вес 80 000—3 000 000, степень кристалличности 75-85 %.
Следует иметь в виду, что названия «полиэтилен низкого давления», «среднего давления», «высокой плотности» и т. д. имеют чисто риторическое значение. Так, полиэтилен, получаемый по 2- и 3-му методам, имеет одинаковую плотность и молекулярный вес. Давление в процессе полимеризации при так называемых низком и среднем давлениях в ряде случаев одно и то же.
Ассортимент полимеров этилена может быть значительно расширен получением сополимеров его с другими мономерами, а также путем получения композиций при компаундировании полиэтилена одного типа с полиэтиленом другого типа, полипропиленом, полиизобутиленом, каучуками и т. п.
На основе полиэтилена и других полиолефинов могут быть получены многочисленные модификации — привитые сополимеры с активными группами, улучшающими адгезию полиолефинов к металлам, окрашиваемость, снижающими его горючесть и т. д.
Особняком стоят модификации так называемого «сшитого» полиэтилена ПЭ-С (PE-X). Суть сшивки состоит в том, что молекулы в цепочке соединяются не только последовательно, но и образуются боковые связи которые соединяют цепочки между собой, за счет этого достаточно сильно изменяются физические и в меньшей степени химические свойства изделий. Различают 4 вида сшитого полиэтилена (по способу производства): пероксидный (а), силановый (b), радиационный (с) и азотный (d). Наибольшее распространение получил РЕх-b, как наиболее быстрый и дешёвый в производстве.
Свойства. Устойчив к действию воды, не реагирует с щелочами любой концентрации, с растворами нейтральных, кислых и основных солей, органическими и неорганическими кислотами, даже концентрированной серной, но разлагается при действии 50%-ой азотной кислоты при комнатной температуре и под воздействием жидкого и газообразного хлора и фтора. При комнатной температуре нерастворим и не набухает ни в одном из известных расворителей. При повышенной температуре (80 °C) растворим в циклогексане и четырёххлористом углероде. Под высоким давлением может быть растворен в перегретой до 180 °C воде. Со временем, деструктирует с образованием поперечных межцепных связей, что приводит к повышению хрупкости на фоне небольшого увеличения прочности. Нестабилизированный полиэтилен на воздухе подвергается термоокислительной деструкции (термостарению). Термостарение полиэтилена проходит по радикальному механизму, сопровождается выделением альдегидов, кетонов, перекиси водорода и др. Полиэтилен низкого давления (HDPE) применяется при строительстве полигонов переработки отходов, накопителей жидких и твердых веществ, способных загрязнять почву и грунтовые воды.
8 Современные физико-химические методы в анализе и идентификации органических соединений
8.1 Спектральные (УФ-, ИК-, ПМР-спектроскопия) и хроматографические (ТСХ, ГЖХ, ВЭЖХ).
УЛЬТРАФИОЛЕТОВАЯ СПЕКТРОСКОПИЯ
(УФ спектроскопия, УФС), раздел оптич. спектроскопии, включающий получение, исследование и применение спектров испускания, поглощения и отражения в ультрафиолетовой области, т. е. в диапазоне длин волн 10-400 нм (волновых чисел 2,5 · 104
- 106
см-1
). УФС при длине волны меньше 185 нм наз. вакуумной, т. к. в этой области УФ излучение настолько сильно поглощается воздухом (гл. обр. кислородом), что необходимо применять вакуумные или наполненные непоглощающим газом спектральные приборы.
Техника измерения УФ спектров в осн. такая же, как спектров в видимой области (см. Спектрофотометрия). Спектральные приборы для УФС отличаются тем, что вместо стеклянных оптич. деталей применяют аналогичные кварцевые (реже флюоритовые или сапфировые), к-рые не поглощают УФ излучение. Для отражения УФ излучения используют алюминиевые покрытия. Приемниками служат обычные или маложелатиновые фотоматериалы, а также фотоэлектрич. приборы, гл. обр. фотоэлектронные умножители, счетчики фотонов, фотодиоды, ионизационные камеры. Для увеличения чувствительности при использовании фотоматериалов иногда регистрируют флуоресценцию, вызванную исследуемым УФ излучением.
Для возбуждения УФ спектров испускания атомов и молекул служат пламя (см. Фотометрия пламени эмиссионная), дуга постоянного или переменного тока, низко- и высоковольтные искры, ВЧ и СВЧ разряд (в т.ч. безэлектродный), плазмотроны, разряд в полом катоде, лазерное излучение и др. (см. Спектральный анализ). УФ спектры поглощения и отражения получают в осн. с использованием таких источников излучения, как дейтериевые (водородные), ртутные, ксеноновые и др. газоразрядные лампы. Используют также нагретые до ок. 3000 К твердые тела, напр. разл. вольфрамовые лампы (с ленточным излучателем или со сферич. анодом, разогреваемым дуговым разрядом, и др.). Источниками линейчатых спектров служат спектральные лампы разл. конструкций (напр., с полым катодом). Применяют также лазеры, излучающие в УФ области (водородный лазер).
Как правило, при облучении УФ излучением B-BO не разрушается и не изменяется, что позволяет получать данные о его хим. составе и структуре. В УФ области проявляются электронные спектры, т. е. положение полос и линий определяется разностью энергий разл. электронных состояний атомов и молекул. Здесь лежат резонансные линии нейтральных, одно- и двукратно ионизованных атомов, а также спектральные линии, испускаемые многократно ионизованными атомами в возбужденном состоянии. В ближней УФ области сосредоточены полосы поглощения большинства полупроводников, возникающие при прямых переходах электронов из валентной зоны в зону проводимости.
В УФ области находятся также электронно-колебат. полосы молекул (колебат. структура проявляется только при низких т-рах; в обычных условиях она приводит к диффузным, т. е. размытым, спектрам), что широко используют в хим. анализе и исследованиях. Появление этих полос связано с переходами электронов между связывающими  и и  , несвязывающими п- и разрыхляющими , несвязывающими п- и разрыхляющими - и - и -орбиталями (см. Молекулярные спектры). Это позволяет использовать УФС для изучения электронного строения молекул, влияния заместителей на хим. св-ва ароматич. соединений, для установления типа хим. связей, определения параметров пов-стей потенц. энергии возбужденных состояний молекул и т. п. В основе этих исследований лежит отнесение полос поглощения УФ спектров к определенным электронным переходам. При этом необходимо учитывать положение и интенсивность полос. Обычно под термином "УФ спектроскопия" понимают именно эту область спектроскопии. -орбиталями (см. Молекулярные спектры). Это позволяет использовать УФС для изучения электронного строения молекул, влияния заместителей на хим. св-ва ароматич. соединений, для установления типа хим. связей, определения параметров пов-стей потенц. энергии возбужденных состояний молекул и т. п. В основе этих исследований лежит отнесение полос поглощения УФ спектров к определенным электронным переходам. При этом необходимо учитывать положение и интенсивность полос. Обычно под термином "УФ спектроскопия" понимают именно эту область спектроскопии.
Для насыщ. углеводородов возможны только  -переходы, требующие больших энергий, и соответствующие им полосы лежат в области вакуумного УФ, напр. в случае метана и этана - при 125 и 135 нм соответственно. Для ненасыщ. соединений характерны -переходы, требующие больших энергий, и соответствующие им полосы лежат в области вакуумного УФ, напр. в случае метана и этана - при 125 и 135 нм соответственно. Для ненасыщ. соединений характерны -переходы, проявляющиеся при длинах волн 165-200 нм. Наличие сопряжения, алкильных или др. заместителей (в т. ч. содержащих гетероатомы) приводит к смещению полос в длинноволновую область (бато-хромный сдвиг), напр. бутадиен поглощает уже при 217 нм. У карбонильных (как и у тиокарбонильных) соед. в наиб. длинноволновой области находится малоинтенсивная полоса, вызванная -переходы, проявляющиеся при длинах волн 165-200 нм. Наличие сопряжения, алкильных или др. заместителей (в т. ч. содержащих гетероатомы) приводит к смещению полос в длинноволновую область (бато-хромный сдвиг), напр. бутадиен поглощает уже при 217 нм. У карбонильных (как и у тиокарбонильных) соед. в наиб. длинноволновой области находится малоинтенсивная полоса, вызванная -переходом, запрещенным по симметрии. В более коротковолновой области проявляются полосы высокой интенсивности -переходом, запрещенным по симметрии. В более коротковолновой области проявляются полосы высокой интенсивности - и - и -переходов. Так, в спектре формальдегида имеются максимумы поглощения при 295 (слабый), 185 и 155 нм. -переходов. Так, в спектре формальдегида имеются максимумы поглощения при 295 (слабый), 185 и 155 нм.
Полосы поглощения сложных эфиров, амидов, галогенан-гидридов смещены в коротковолновую область, а полосы тиокарбонильных соед.- в длинноволновую область по сравнению с полосами поглощения соответствующих карбонильных соед., напр.: максимумы поглощения CH3
C(O)H, CH3
C(O)NH2
и CH3
C(S)NH2
наблюдаются при 290, 214 и 358 нм соответственно. Вследствие гибридизации неподеленной пары электронов азота в соед., содержащих группу C = N, интенсивность полосы  -перехода у них выше, чем у карбонильных соединений. В спектрах нитросоед. положение и интенсивность полосы -перехода у них выше, чем у карбонильных соединений. В спектрах нитросоед. положение и интенсивность полосы -перехода зависят от природы соседнего с нитрогруппой атома. Так, у О-нитросоед. эта малоинтенсивная полоса расположена в более коротковолновой области, чем у С-нитросоединений. В спектре нитрами-нов (N-NO2
) полоса -перехода зависят от природы соседнего с нитрогруппой атома. Так, у О-нитросоед. эта малоинтенсивная полоса расположена в более коротковолновой области, чем у С-нитросоединений. В спектре нитрами-нов (N-NO2
) полоса -перехода наиб. интенсивная. -перехода наиб. интенсивная.
Для азо- и нитрозосоединений также характерны  -переходы. Полосы УФ спектра N- и О-нитрозосоединений смещены в коротковолновую область по сравнению с полосами С-нитрозосоединений. Сопряжение кратных связей с такими азотсодержащими хромофорными группами, как NO2
, NO, N = N, N3
, вызывает батохромный сдвиг всех полос поглощения и возрастание их интенсивности. -переходы. Полосы УФ спектра N- и О-нитрозосоединений смещены в коротковолновую область по сравнению с полосами С-нитрозосоединений. Сопряжение кратных связей с такими азотсодержащими хромофорными группами, как NO2
, NO, N = N, N3
, вызывает батохромный сдвиг всех полос поглощения и возрастание их интенсивности.
Характер спектра поглощения зависит от взаимного расположения хромофоров. Если хромофорные группы соединены непосредственно, то в спектре наблюдаются сильные изменения по сравнению со спектрами соед. с изолированными хромофорными группами. Относит. расположение хромофоров у кратных связей позволяет различать цис- и транс-изомеры.
Полосы в спектрах ароматич. соед. связаны с переходами  -электронов ароматич. системы. На вид спектра влияют заместители: такие как алкил, галогены - незначительно, группы с неподеленными парами электронов (ОН, OR, NH2
, NF2
) - сильно. Если имеются карбонильная, нитро- или нитрозогруппа, то в спектре дополнительно наблюдаются полосы -электронов ароматич. системы. На вид спектра влияют заместители: такие как алкил, галогены - незначительно, группы с неподеленными парами электронов (ОН, OR, NH2
, NF2
) - сильно. Если имеются карбонильная, нитро- или нитрозогруппа, то в спектре дополнительно наблюдаются полосы  -перехода. В спектрах нек-рых замещенных бензола, напр. нитробензола, удается выделить полосы с внутримол. переносом заряда (соответствующие переходам, при к-рых происходит преимуществ, уменьшение электронной плотности на одном участке молекулы и ее увеличение на др. участке). -перехода. В спектрах нек-рых замещенных бензола, напр. нитробензола, удается выделить полосы с внутримол. переносом заряда (соответствующие переходам, при к-рых происходит преимуществ, уменьшение электронной плотности на одном участке молекулы и ее увеличение на др. участке).
УФ спектры ароматич. соед. зависят не только от характера, но и от взаимного расположения заместителей, так, в спектрах орто- и меmа-нитроанилина имеются три полосы, вызванные переносом заряда от донора к акцептору, от кольца к акцептору и локальным возбуждением бензольного кольца с вкладом переноса заряда от донора к кольцу. пара-Изомер имеет те же переходы, но из-за совпадения направления переноса заряда во всех трех случаях в спектре появляется одна интенсивная полоса поглощения (при 320 нм).
Насыщ. гетероциклы имеют полосы, соответствующие  -переходам. Полосы поглощения кислород- и азотсодержащих соед. лежат в области вакуумного УФ. Серосодер-жащие соед. имеют соответствующие полосы в обычной УФ области. -переходам. Полосы поглощения кислород- и азотсодержащих соед. лежат в области вакуумного УФ. Серосодер-жащие соед. имеют соответствующие полосы в обычной УФ области.
Замена в ароматич. кольце группы =СН на =N приводит к повышению интенсивности длинноволновой полосы поглощения и появлению полосы  -перехода (к-рая в случае пиридина проявляется только в спектрах его паров). По мере увеличения числа атомов N в цикле полосы -перехода (к-рая в случае пиридина проявляется только в спектрах его паров). По мере увеличения числа атомов N в цикле полосы  -переходов сдвигаются в длинноволновую область. -переходов сдвигаются в длинноволновую область.
Наличие интенсивных характеристич. полос в УФ спектрах мн. хим. соединений используется для разработки методов их идентификации и количеств, определения. Последние основаны на законе Бугера-Ламберта-Бера (см. Абсорбционная спектроскопия) и отличаются селективностью и высокой чувствительностью - до 10-7
% по массе. Имеются хим. сенсоры со световодами, измеряющие поглощение определяемого в-ва в УФ области.
УФС применяют также для изучения кинетики хим. и фотохим. р-ций, исследования люминесценции, уровней энергии и вероятностей квантовых переходов в твердых телах и т. д. Особое значение имеет УФС для установления состава космич. объектов и изучения протекающих на них процессов.
ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
(ИК спектроскопия), раздел мол. оптич. спектроскопии, изучающий спектры поглощения и отражения электромагн. излучения в ИК области, т.е. в диапазоне длин волн от 106
до 103
м. В координатах интенсивность поглощенного излучения - длина волны (или волновое число) ИК спектр представляет собой сложную кривую с большим числом максимумов и минимумов. Полосы поглощения появляются в результате переходов между колебат. уровнями осн. электронного состояния изучаемой системы (см. Колебательные спектры). Спектральные характеристики (положения максимумов полос, их полуширина, интенсивность) индивидуальной молекулы зависят от масс составляющих ее атомов, геом. строения, особенностей межатомных сил, распределения заряда и др. Поэтому ИК спектры отличаются большой индивидуальностью, что и определяет их ценность при идентификации и изучении строения соединений. Для регистрации спектров используют классич. спектрофотометры и фурье-спектрометры. Осн. части классич. спектрофотометра - источник непрерывного теплового излучения, монохроматор, неселективный приемник излучения. Кювета с в-вом (в любом агрегатном состоянии) помещается перед входной (иногда за выходной) щелью. В качестве диспергирующего устройства монохроматора применяют призмы из разл. материалов (LiF, NaCl, KCl, CsF и др.) и дифракц. решетки. Последовательное выведение излучения разл. длин волн на выходную щель и приемник излучения (сканирование) осуществляется поворотом призмы или решетки. Источники излучения - накаливаемые электрич. током стержни из разл. материалов. Приемники: чувствительные термопары, металлич. и полупроводниковые термосопротивления (болометры) и газовые термопреобразователи, нагрев стенки сосуда к-рых приводит к нагреву газа и изменению его давления, к-рое фиксируется. Выходной сигнал имеет вид обычной спектральной кривой. Достоинства приборов классич. схемы: простота конструкции, относит. дешевизна. Недостатки: невозможность регистрации слабых сигналов из-за малого отношения сигнал: шум, что сильно затрудняет работу в далекой ИК области; сравнительно невысокая разрешающая способность (до 0,1 см1
), длительная (в течение минут) регистрация спектров. В фурье-спектрометрах отсутствуют входная и выходная щели, а осн. элемент - интерферометр. Поток излучения от источника делится на два луча, к-рые проходят через образец и интерферируют. Разность хода лучей варьируется подвижным зеркалом, отражающим один из пучков. Первоначальный сигнал зависит от энергии источника излучения и от поглощения образца и имеет вид суммы большого числа гармонич. составляющих. Для получения спектра в обычной форме производится соответствующее фурье-преобразование с помощью встроенной ЭВМ. Достоинства фурье-спектрометра: высокое отношение сигнал : шум, возможность работы в широком диапазоне длин волн без смены диспергирующего элемента, быстрая (за секунды и доли секунд) регистрация спектра, высокая разрешающая способность (до 0,001 см1
). Недостатки: сложность изготовления и высокая стоимость. Все спектрофотометры снабжаются ЭВМ, к-рые производят первичную обработку спектров: накопление сигналов, отделение их от шумов, вычитание фона и спектра сравнения (спектра р-рителя), изменение масштаба записи, вычисление эксперим. спектральных параметров, сравнение спектров с заданными, дифференцирование спектров и др. Кюветы для ИК спектрофотометров изготовляют из прозрачных в ИК области материалов. В качестве р-рителей используют обычно ССl4
, СНСl3
, тетрахлорэтилен, вазелиновое масло. Твердые образцы часто измельчают, смешивают с порошком КВr и прессуют таблетки. Для работы с агрессивными жидкостями и газами применяют спец. защитные напыления (Ge, Si) на окна кювет. Мешающее влияние воздуха устраняют вакуумированием прибора или продувкой его азотом. В случае слабо поглощающих в-в (разреженные газы и др.) применяют многоходовые кюветы, в к-рых длина оптич. пути достигает сотен метров благодаря многократным отражениям от системы параллельных зеркал. Большое распространение получил метод матричной изоляции, при к-ром исследуемый газ смешивают с аргоном, а затем смесь замораживают. В результате полуширина полос поглощения резко уменьшается и спектр получается более контрастным. Применение спец. микроскопич. техники позволяет работать с объектами очень малых размеров (доли мм). Для регистрации спектров пов-сти твердых тел применяют метод нарушенного полного внутр. отражения. Он основан на поглощении поверхностным слоем в-ва энергии электромагн. излучения, выходящего из призмы полного внутр. отражения, к-рая находится в оптич. контакте с изучаемой пов-стью. Инфракрасную спектроскопию широко применяют для анализа смесей и идентификация чистых в-в. Количеств. анализ основан на законе Бугера-Ламберта-Бера (см. Абсорбционная спектроскопия), т. е. на зависимости интенсивности полос поглощения от концентрации в-ва в пробе. При этом о кол-ве в-ва судят не по отд. полосам поглощения, а по спектральным кривым в целом в широком диапазоне длин волн. Если число компонентов невелико (4-5), то удается математически выделить их спектры даже при значит. перекрывании последних. Погрешность количеств. анализа, как правило, составляет доли процента. Идентификация чистых в-в производится обычно с помощью информационно-поисковых систем путем автоматич. сравнения анализируемого спектра со спектрами, хранящимися в памяти ЭВМ. Характерные области поглощения ИК излучения наиб. часто встречающихся функц. групп хим. соед. приведены в табл. на форзаце в конце тома. Для идентификации новых в-в (молекулы к-рых могут содержать до 100 атомов) применяют системы искусств. интеллекта. В этих системах на основе спектроструктурных корреляций генерируются мол. структуры, затем строятся их теоретич. спектры, к-рые сравниваются с эксперим. данными. Исследование строения молекул и др. объектов методами инфракрасной спектроскопии подразумевает получение сведений о параметрах мол. моделей и математически сводится к решению т. наз. обратных спектральных задач. Решение таких задач осуществляется последовательным приближением искомых параметров, рассчитанных с помощью спец. теории спектральных кривых к экспериментальным. Параметрами мол. моделей служат массы составляющих систему атомов, длины связей, валентные и торсионные углы, характеристики потенциальной пов-сти (силовые постоянные и др.), дипольные моменты связей и их производные по длинам связей и др. Инфракрасная спектроскопия позволяет идентифицировать пространственные и конформационные изомеры, изучать внутри- и межмолекулярные взаимод., характер хим. связей, распределение зарядов в молекулах, фазовые превращения, кинетику хим. р-ций, регистрировать короткоживущие (время жизни до 106
с) частицы, уточнять отдельные геом. параметры, получать данные для вычисления термодинамич. ф-ций и др. Необходимый этап таких исследований - интерпретация спектров, т.е. установление формы нормальных колебаний, распределения колебат. энергии по степеням свободы, выделение значимых параметров, определяющих положение полос в спектрах и их интенсивности. Расчеты спектров молекул, содержащих до 100 атомов, в т.ч. полимеров, выполняются с помощью ЭВМ. При этом необходимо знать характеристики мол. моделей (силовые постоянные, электрооптич. параметры и др.), к-рые находят решением соответствующих обратных спектральных задач или квантовохим. расчетами. И в том, и в другом случае обычно удается получать данные для молекул, содержащих атомы лишь первых четырех периодов периодич. системы. Поэтому инфракрасная спектроскопия как метод изучения строения молекул получил наиб. распространение в орг. и элементоорг. химии. В отд. случаях для газов в ИК области удается наблюдать вращат. структуру колебат. полос. Это позволяет рассчитывать дипольные моменты и геом. параметры молекул, уточнять силовые постоянные и т.д.
Применение спектроскопии ЯМР (ПМР).
Спектроскопия ЯМР относится к неразрушающим методам анализа. Совр. импульсная ЯМР фурье-спектроскопия позволяет вести анализ по 80 магн. ядрам. ЯМР спектроскопия - один из осн. физ.-хим. методов анализа, ее данные используют для однозначной идентификации как промежут. продуктов хим. р-ций, так и целевых в-в. Помимо структурных отнесений и количеств. анализа, спектроскопия ЯМР приносит информацию о конформационных равновесиях, диффузии атомов и молекул в твердых телах, внутр. движениях, водородных связях и ассоциации в жидкостях, кето-енольной таутомерии, металлo- и прототропии, упорядоченности и распределении звеньев в полимерных цепях, адсорбции в-в, электронной структуре ионных кристаллов, жидких кристаллов и др. Спектроскопия ЯМР - источник информации о структуре биополимеров, в т. ч. белковых молекул в р-рах, сопоставимой по достоверности с данными рентгеноструктурного анализа. В 80-е гг. началось бурное внедрение методов спектроскопии и томографии ЯМР в медицину для диагностики сложных заболеваний и при диспансеризации населения.
Число и положение линий в спектрах ЯМР однозначно характеризуют все фракции сырой нефти, синтетич. каучуков, пластмасс, сланцев, углей, лекарств, препаратов, продукции хим. и фармацевтич. пром-сти и др.
Интенсивность и ширина линии ЯМР воды или масла позволяют с высокой точностью измерять влажность и масличность семян, сохранность зерна. При отстройке от сигналов воды можно регистрировать содержание клейковины в каждом зерне, что так же, как и анализ масличности, позволяет вести ускоренную селекцию с.-х. культур.
Применение все более сильных магн. полей (до 14 Тл в серийных приборах и до 19 Тл в эксперим. установках) обеспечивает возможность полного определения структуры белковых молекул в р-рах, экспресс-анализа биол. жидкостей (концентрации эндогенных метаболитов в крови, моче, лимфе, спинномозговой жидкости), контроля качества новых полимерных материалов. При этом применяют многочисленные варианты многоквантовых и многомерных фурье-спектроскопич. методик.
8.2 Масс-спектрометрический метод
Применение масс-спектрометрии. Масс-спектрометрию широко применяют в разл. областях науки и техники: в химии и нефтехимии, физике, геологии, биологии, медицине, в пром-сти полимеров, в лакокрасочной и хим. пром-сти, в произ-ве полупроводников и сверхчистых материалов, в ядерной технике, в с. х-ве и ветеринарии, в пищ. пром-сти, при анализе продуктов загрязнения окружающей среды и мн. др. Большие успехи достигнуты при анализе биологически важных в-в; показана возможность структурного анализа полисахаридов с мол. м. до 15000, белков с мол. м. до 45000 и т.д. Масс-спектрометрия нашла применение как экспрессный метод газового анализа в медицине; принципы масс-спектрометрии лежат в основе устройства наиб. чувствит. течеискателей. Отечеств. масс-спектрометры, выпускаемые для разл. целей, имеют индексы: для исследования изотопного состава - МИ, для исследования хим. состава - MX, для структурного анализа - МС. Macс-спектрометрия в органической химии позволяет измерить точную мол. массу и рассчитать элементный состав исследуемого в-ва, установить хим. и пространств. строение, определить изотопный состав, провести качеств. и количеств. анализ сложных смесей орг. соединений. Одна из важнейших задач - нахождение зависимости между характером масс-спектра и строением исследуемой орг. молекулы. При ионизации орг. молекулы образуется мол. ион, в к-ром далее происходят процессы гетеро- и гомолитич. разрыва связей или разрыва связей с перегруппировкой молекулы и образование осколочных ионов, к-рые в свою очередь могут подвергаться дальнейшему распаду. Последоват. распады ионов, устанавливаемые из масс-спектра, наз. направлениями или путями распада. Направления распада - важная характеристика каждого класса соединений. Совокупность всех направлений распада составляет характерную для каждого орг. соед. схему фрагментации. Если масс-спектр прост, схема фрагментации сводится к одному пути распада, напр. при распаде мол. иона СН3ОН+ последовательно образуются ионы СН2=ОН+ и Н—С=О+. В случае сложных масс-спектров схема фрагментации отвечает многим, часто перекрывающимся направлениям распада, напр. схема фрагментации полипептида:
Мол. ион пептида распадается в результате разрыва связей СН—СО, СО—NH, NH—СН и СН—R с образованием осколочных ионов соотв. Аn и Хn, Вn и Yn, Сn и Zn, Sn и Rn (n - номер аминокислотного остатка в пептидной цепи), к-рые далее распадаются таким же образом. Общее кол-во пиков ионов в таком спектре может достигать неск. сотен. Кол-во фрагментов определяется строением исследуемой молекулы, запасом внутр. энергии мол. и осколочных ионов и промежутком времени между образованием иона и его детектированием. Поэтому при интерпретации масс-спектров необходимо учитывать как условия измерений (энергию ионизирующих электронов, ускоряющее напряжение, давление паров в ионном источнике, т-ру ионизац. камеры), так и конструктивные особенности прибора. При макс. стандартизации условий измерений удается получать достаточно воспроизводимые масс-спектры. Сравнение масс-спектра исследуемой системы со спектром, имеющимся в каталоге, -наиб. быстрый и простой способ структурного анализа, идентификации в-в при определении загрязнения окружающей среды, контроле продуктов питания человека и животных, изучении процессов метаболизма лек. препаратов, в криминалистике и т.д. Однако идентификация лишь на основании масс-спектра не может быть однозначной, напр. не все изомерные в-ва образуют различающиеся масс-спектры. В условиях масс-спектрометрии часть возбужденных ионов распадается после выхода из ионного источника. Такие ионы наз. метастабильными. В масс-спектрах они характеризуются уширенными пиками при нецелочисленных значениях т/z. Один из методов изучения таких ионов - спектроскопия масс и кинетич. энергий ионов. Изучение распада метастабильных ионов проводят на приборах, у к-рых магн. анализатор предшествует электрическому. Магн. анализатор настраивают таким образом, чтобы он пропустил метастабильный ион, к-рый при определенном напряжении на электрич. анализаторе проходит в детектор. Если такой ион распадается в пространстве между анализаторами, то образующиеся вторичные ионы не могут пройти через электрич. анализатор при установленном напряжении из-за недостатка энергии. Для попадания вторичных ионов в детектор изменяют напряжение электрич. анализатора. Это напряжение связано с массой вторичного иона соотношением m2 = Е2m*/Е0, где m* - метастабильный ион, m2 - вторичный ион, Е0 и Е2 - начальное и конечное напряжение электрич. анализатора. Таким образом определяются массы всех ионов, образующихся при распаде метастабильных ионов и устанавливаются тем самым схемы их фрагментации. Если в области между двумя анализаторами создать область повыш. давления (установить камеру столкновений, заполненную инертным газом), то в результате соударений ионов с молекулами газа их внутр. энергия будет увеличиваться и, следовательно, увеличится вероятность образования вторичных ионов. Такой метод, наз. тандемным, используют для структурного анализа индивидуальных компонентов сложных смесей без предварит. разделения. Наряду со структурными исследованиями масс-спектрометрию применяют для количеств. анализа орг. в-в. Количеств. анализ основан на определении интенсивностей пиков ионов с определенным значением т/z. Его проводят хромато-масс-спектрометрически (см. Хромато-масс-спектрометрия) или в системе прямого ввода. Для повышения точности определения применяют внутр. стандарты, в качестве к-рых используют меченые соед. или соед. близкие по строению к исследуемым, напр. гомологи. В последнем случае необходимо построение калибровочных кривых. Измерение содержания исследуемого в-ва проводят с учетом кол-ва добавляемого стандарта по отношению площадей пиков, соответствующих определяемому в-ву и внутр. стандарту. Погрешность метода b7%, предел определения 0,01 мкг/мл. Лучшие результаты дает применение меченых соед.; при этом отпадает необходимость в построении калибровочных кривых. Количеств. определение труднолетучих в-в проводят в системе прямого ввода, детектируя их по одному или неск. ионам, характерным для исследуемого соединения. По мере плавного повышения т-ры испарителя происходит испарение и частичное фракционирование исследуемых в-в. Т. обр., для каждого в-ва получают кривую испарения, площадь под к-рой прямо пропорциональна кол-ву соед., внесенного в масс-спектрометр. Абс. чувствительность метода, наз. методом интегрирования ионного тока, 10-7 г. Достоинство метода - отсутствие необходимости предварит. очистки исследуемых в-в. При исследовании соед. с электроф. группировками, изомерных орг. молекул, полимеров, азокрасителей, биологически активных в-в применяют масс-спектрометрию отрицательно заряженных ионов. Эти ионы обладают меньшим запасом внутр. энергии, чем положительно заряженные ионы, поэтому в масс-спектрах дают интенсивные пики мол. ионов и малое кол-во осколочных ионов.
9 Основные приёмы техники лабораторной работы (экстракция, выделение, очистка, определение температуры плавления, различные виды перегонки, кристаллизация)
ВЫДЕЛЕНИЕ. Синтезируемое вещество, получаемое в результате какой-либо реакции, обычно находится в реакционной смеси совместно с другими веществами (другие продукты, получающиеся по основному уравнению реакции; побочные продукты реакции; растворитель, в котором проводилась реакция). Поэтому всегда возникает задача выделения нужного вещества из весьма сложной подчас смеси. Иногда такое выделение удается не сразу; часто вначале вещество выделяют не вполне чистым и только в результате дальнейшей обработки получают чистый продукт.
Между методами выделения вещества из сложной реакционной смеси и методами его последующей очистки нет резкой разницы. Обычно и в том и в другом случаях используют различие в растворимости и в летучести веществ, имеющихся в смеси. Кроме того, для очистки пользуются различной способностью разных веществ поглощаться адсорбентами, например активным углем.
Использование различия в растворимости органических веществ лежит в основе выделения и очистки их методами кристаллизации и экстракции, а различия в летучести - в основе очистки перегонкой.
Кристаллизация
При очистке органического вещества кристаллизацией задача заключается в том, чтобы создать благоприятные условия для выделения данного вещества в кристаллическом состоянии из пересыщенного раствора и в то же время удержать в растворе сопутствующие примеси.
Из двух методов получения пересыщенных растворов - путем испарения части растворителя и путем охлаждения растворов, насыщенных при нагревании, - предпочитают пользоваться последним. При кристаллизации через охлаждение пользуются такими растворителями, в которых растворимость кристаллизуемого вещества резко изменяется с температурой. Существенной является также способность растворителя хорошо растворять примеси; чем больше разница в величинах растворимости основного продукта и примесей, тем легче осуществляется очистка. Нужно отметить, что загрязнения могут сильно влиять на скорость кристаллизации и на полноту выделения кристаллизуемого вещества из раствора. Иногда в присутствии значительного количества примесей кристаллизация может вообще не наступить, а если и удается добиться выделения кристаллов, то потери вещества в маточном растворе оказываются слишком большими. Поэтому во многих случаях к очистке вещества путем кристаллизации следует прибегать лишь после освобождения его от значительной части примесей другими способами, например перегонкой.
В качестве растворителя при кристаллизации наиболее часто применяют воду, этиловый спирт, метиловый спирт, бензин, бензол, петролейный эфир, этиловый эфир, уксусноэтиловый эфир, ледяную уксусную кислоту, хлороформ. Для труднорастворимых соединений используют также нитробензол, пиридин, фенол, анилин.
Большое значение для успеха работы имеет правильный выбор растворителя. При выборе растворителя необходимо учитывать состав и строение растворяемого вещества. Так, вещества, содержащие гидроксильные группы, в большинстве случаев более или менее хорошо растворяются в воде. Увеличение длины углеводородной цепи, например в высших спиртах, резко уменьшает растворимость в воде, но увеличивает растворимость в спиртах и углеводородах.
Окончательный выбор растворителя можно произвести лишь опытным путем. Для этого берут несколько пробирок, помещают в них небольшое количество вещества (например, по 0,2 г), прибавляют 0,5-1 мл различных растворителей и нагревают до полного растворения. Наиболее подходящим будет тот растворитель, из которого по охлаждении выделяется хорошо образованные кристаллы в небольшом количестве. Если в одном из растворителей вещество растворяется очень хорошо, а в другом - плохо, то следует испытать их смесь. Часто применяют смесь спирта с водой, ацетона с водой, эфира с бензолом.
Растворимость вещества в выбранном растворителе на холоду не должна быть слишком большой, так как это приводит к чрезмерно большим потерям вещества в маточном растворе. Кроме того, в этом случае пришлось бы работать с небольшими объемами жидкости, что привело бы к увеличению механических потерь (размазывание по стенкам, неполнота стекания и т. п.). В случае малой растворимости работа осложняется необходимостью оперировать со слишком большими объемами растворов.
Самая кристаллизация проводится следующим образом. Подлежащие очистке вещество помещают в колбу, обливают небольшим количеством растворителя, нагревают до кипения и затем добавляют понемногу новые порции растворителя (доводя после этого раствор снова до кипения) до полного растворения вещества*1. Чтобы растворитель не испарялся, колбу соединяют с обратным холодильником и растворитель приливают через трубку холодильника. Нагревание обычно ведут на водяной бане, за исключением тех случаев, когда работают с высококипящими растворителями; при приливании горючих растворителей горелку отставляют.
Полученный концентрированный раствор необходимо профильтровать (для удаления нерастворимых примесей, волокон фильтровальной бумаги и других загрязнений). Фильтрование ведут с отсасыванием через достаточно большую воронку Бюхнера (Рис. 7), вставленную в толстостенную коническую колбу для отсасывания*2. Если вещество при охлаждении кристаллизуется очень легко, то в случае концентрированных растворов кристаллизация начинается в самой воронке, ее отверстия забиваются и фильтрование прекращается. Чтобы избежать этого, растворитель берут в избытке (небольшом), а воронку перед фильтрованием осторожно подогревают пламенем горелки.
Во избежание кристаллизации во время фильтрования можно также пользоваться воронкой для горячего фильтрования. Эта воронка имеет двойные стенки, между которыми наливается вода, подогреваемая горелкой. Внутрь этой воронки вставляется обычная стеклянная воронка с фильтром.
При работе с легколетучими растворителями фильтрование с отсасыванием приводит к слишком большим потерям растворителя за счет испарения. В этих случаях следует фильтровать через обычную коническую воронку со вставленным в нее складчатым фильтром из неплотной фильтровальной бумаги; для уменьшения испарения растворителя воронку накрывают часовым стеклом (выпуклой стороной книзу).
Для получения хорошо образованных кристаллов необходимо охлаждать раствор медленно, при полном покое. Часто при попадании горячего фильтруемого раствора в холодный приемник наблюдается быстрое выделение обычно плохо образованных кристаллов. В этом случае профильтрованный раствор необходимо снова нагреть до растворения кристаллов и оставить медленно охлаждаться. Во многих случаях кристаллизация наступает очень медленно. Для ускорения ее прибегают к трению стеклянной палочкой о стенки сосуда или к внесению "затравки" (кристаллик ранее полученного препарата того же вещества). Как только кристаллизация начнется, раствор оставляют стоять в покое.
Для более полного выделения кристаллов из маточного раствора часто прибегают к его охлаждению при помощи охлаждающих смесей или же ставят сосуд с раствором в холодильный шкаф. Растворимость большинства веществ при низких температурах уменьшается, и поэтому путем охлаждения достигается большая полнота выделения кристаллизуемого вещества из раствора. Однако нужно учитывать, что понижение температуры может уменьшить скорость роста кристаллов, что особенно заметно в случае вязких жидкостей.
Для удобства извлечения образовавшихся кристаллов рекомендуется проводить кристаллизацию в конических колбах или в стаканах, но не в обычных плоскодонных колбах. При работе с летучими растворителями пользуются только коническими колбами, которые во избежание испарения растворителя накрывают часовым стеклом (выпуклой стороной кверху). Ни в коем случае не следует колбу с горячим раствором плотно закрывать пробкой: при охлаждении в колбе создается вакуум (вследствие конденсации паров) и она может быть раздавлена атмосферным давлением.
Для удаления из раствора окрашенных и смолообразных примесей, затрудняющих кристаллизацию и загрязняющих получаемые кристаллы, с успехом применяют активный уголь (крупнопористые сорта). Уголь, во избежание внезапного вскипания жидкости, следует вносить в несколько охлажденный раствор, когда все подлежащее кристаллизации вещество растворилось. После прибавления активного угля раствор нагревают до кипения, кипятят несколько минут и затем фильтруют.
Уголь прибавляют в количестве, необходимом для полного обесцвечивания раствора, избегая в то же время большого избытка. Для этого уголь вносят небольшими порциями, после внесения каждой из них раствор кипятят и затем дают ему несколько отстояться, чтобы можно было установить, в достаточной ли мере удалены смолистые и окрашенные примеси. Так поступают до тех пор, пока не будет достигнут нужный эффект очистки.
Иногда частицы слишком мелко растертого угля проходят сквозь фильтр и загрязняют фильтрат. Этот недостаток может быть устранен предварительным взмучиванием угля в воде и декантацией (после отстаивания) взвешенный мелких частиц. При работе с неводными растворителями промытый уголь высушивают на водяной бане.
Если раствор фильтруется плохо и фильтр забивается, то иногда полезно прибавить к углю немного мелких древесных опилок. В тех случаях, когда после осветления углем вещество предполагают подвергнуть анализу (элементарному), нужно особенно тщательно следить, чтобы частицы угля не попали в фильтрат. Лучше всего перед анализом перекристаллизовать вещество еще раз, уже без применения активного угля.
Полученные кристаллы отделяют от маточного раствора фильтрованием с отсасыванием на воронке Бюхнера или, в случае жидкостей, действующих на бумагу, - на воронках с фильтровальными пластинками из пористого стекла. Размеры воронки должны соответствовать количеству отсасываемого вещества; применение воронок слишком больших размеров приводит к ненужным потерям вещества. Для отфильтровывания очень малых количеств кристаллов (порядка 0,1 г и менее) пользуются обычной маленькой стеклянной воронкой, в которую вставляют стеклянную палочку с расплюснутым концом - "пуговкой". Для приготовления такой "пуговки" конец тонкой стеклянной палочки нагревают до размягчения и затем прижимают ко дну ступки, к керамиковой плитке и т. п. Стеклянная палочка должна быть настолько тонкой и длинной, чтобы она свободно входила в трубку воронки и конец ее выдавался немного снизу. На "пуговку" кладут кружок фильтровальной бумаги немного большего диаметра, так чтобы он плотно прилегал к стенкам воронки (рис. 8). Воронку вставляют или в маленькую колбу для отсасывания, или в укрепленную в штативе пробирку для отсасывания.
Для того чтобы фильтр плотно прилегал к стенкам воронки, его полезно смочить водой, отсосать воду, промыть небольшим количеством спирта и под конец - тем растворителем, который нужно будет отсасывать.
Фильтр, вкладываемый в воронку Бюхнера, должен быть несколько меньшего диаметра, чем воронка, и, полностью закрывая все отверстия дна воронки, не должен в то же время прилегать к ее стенкам. Перед фильтрованием фильтр нужно смочить растворителем и затем включить насос. Кристаллы из сосуда, в котором производилась кристаллизация, переносят на фильтр с помощью стеклянной палочки. Кристаллы, приставшие к стенкам сосуда, смывают небольшими порциями отфильтрованного маточного раствора. Для более полного удаления маточного раствора часто бывает полезным отжать кристаллы на фильтре (не прекращая отсасывания) при помощи шпателя, пестика или стеклянной пробки.
После того как маточный раствор отфильтрован, не следует просасывать воздух через кристаллы, так как растворитель при этом испаряется и содержащиеся в нем примеси остаются на кристаллах. Для удаления маточного раствора, захваченного кристаллами, их необходимо промыть возможно малым количеством холодного растворителя. Для этого перекрывают отсасывание, смачивают осадок растворителем, дают немного постоять, чтобы осадок равномерно пропитался жидкостью, и отсасывают. Эту операцию повторяют еще раз или два (но не более). Большинство органических веществ довольно хорошо растворяется даже в холодных растворителях; поэтому хорошее промывание осадка при минимальных потерях вещества, требует от работающего известного навыка.
В маточных растворах и промывных жидкостях часто остается такое количество вещества, которым не следует пренебрегать. В таких случаях надо отогнать часть растворителя и снова довести раствор до кристаллизации. Полученные при этом кристаллы обычно бывают менее чистыми, чем первая порция, и их следует перекристаллизовать еще раз.
Высушивание осадка. По окончании промывания осадок вместе с фильтром вынимают из воронки, кладут на сложенную в несколько раз фильтровальную бумагу, удаляют пинцетом фильтр и отжимают осадок между листьями фильтровальной бумаги. В большинстве случаев для окончательного удаления растворителя оказывается достаточным простое высушивание осадка на воздухе при комнатной температуре. С этой целью отжатый осадок рассыпают тонким слоем на листе фильтровальной бумаги, покрывают (для защиты от пыли) другим листом фильтровальной бумаги и оставляют до полного высыхания.
Иногда высушивание препарата можно ускорить, нагревая его в сушильном шкафу. Этот способ следует, однако, применять с осторожностью и только в случае вещества с высокой температурой плавления, так как небольшая примесь еще не удаленного растворителя может существенно снизить температуру плавления и вещество может при нагревании расплавиться.
Вещества гигроскопические нужно сушить в эксикаторе. В качестве водуотнимающих средств в эксикатор помещают окись алюминия, хлористый кальций, концентрированную серную кислоту или фосфорный ангидрид. Следует особенно рекомендовать применение окиси алюминия и хлористого кальция.
Окись алюминия очень энергично поглощает воду и может связать до 15-20% воды от собственного веса. Она легко регенерируется путем нагревания до 175° в течение 6 час. с последующим охлаждением в эксикаторе. Хлористый кальций несколько уступает окиси алюминия (а также и серной кислоте) по способности связывать воду, но он является легко доступным, дешевым продуктом, легко регенерируется путем прокаливания и свободен от тех недостатков, которые, как указано ниже, присущи серной кислоте.
Серная кислота, хорошо поглощая воду, одновременно поглощает и пары органических веществ; в результате их постепенного окисления она восстанавливается до сернистого ангидрида, который может реагировать с высушиваемом веществом. Другим недостатком применения серной кислоты является возможность ее расплескивания при переноске эксикатора, в результате чего брызги кислоты могут падать на дно сосуда с высушиваемым веществом. Чтобы кислота не расплескивалась, на дно эксикатора насыпают достаточно толстым слоем битое стекло. Для того чтобы установить момент, когда серная кислота станет непригодной в качестве высушивающего средства, в ней растворяют (перед помещением в эксикатор) сернокислый барий (из расчета 18 г сернокислого бария на 1 л концентрированной серной кислоты). Выпадение осадка сернокислого бария указывает на то, что кислота уже непригодна для сушки и должна быть заменена свежей. Нужно отметить, что при вакууме порядка 1 мм серная кислота несколько летуча и поэтому ее не рекомендуется применять в вакуум-эксикаторах при больших разрежениях.
Фосфорный ангидрид связывает воду очень энергично, но при этом на его поверхности образуется сиропообразная корочка, препятствующая дальнейшему поглощению воды, что является существенным недостатком.
Определение температуры плавления
Температурой плавления считается температура, при которой замечается первое появление жидкой фазы. Разность между температурой, при которой появляется жидкая фаза, и температурой полного расплавления вещества, не должна превышать 0,5 °С. Незначительные загрязнения вещества иногда сильно понижают температуру его плавления, и оно происходит в более широком интервале температур. Такое явление используют для установления идентичности двух веществ с одинаковой температурой плавления. Для этого смешивают равные количества двух веществ. Если температура плавления этой "смешанной" пробы остается неизменной, то делают заключение об идентичности обоих веществ. Понижение же температуры плавления пробы служит признаком неидентичности. Оценка идентичности исследуемого вещества по температуре плавления ''смешанной" пробы является настолько общепринятой, что этот прием считается достаточным для вынесения окончательного решения.
Многие органические вещества плавятся с разложением, которое обычно обнаруживается по окрашиванию расплава или выделению газа. В качестве характеристики веществ, которые плавятся с разложением, в справочнике приведена величина температуры плавления с дополнением "разл.". Существуют различные приборы для определения температуры плавления органических веществ.
Наиболее простой прибор для определения температуры плавления состоит из круглодонной колбы, заполненной соответствующей обогревающей жидкостью и имеющей боковые отверстия для испарения этой жидкости. В колбу вставлена пробирка с термометром, к которому прикреплен капилляр с веществом.
В качестве теплопередающей среды используют воду, серную кислоту, силиконовое масло и др. В данном приборе температуру плавления органического кристаллического вещества определяют в капилляре, запаянном с одного конца. Испытуемое вещество растирают в ступке. Открытым концом капилляра набирают в него немного вещества и бросают его запаянным концом вниз в стеклянную трубку длиной 60-80 см, поставленную вертикально на лабораторный стол. Эту операцию наполнения повторяют несколько раз до получения в капилляре хорошо уплотненного столбика вещества высотой 3-4 мм. Наполненный капилляр закрепляют резиновым кольцом на термометре так, чтобы проба вещества находилась на уровне ртутного шарика термометра. Нагревают прибор электрической плиткой. Когда исследуемое вещество начинает заметно плавиться либо сжиматься и мокнуть, плитку убирают. Началом плавления считают появление первой капли в капилляре, а окончанием – исчезновение последних кристалликов вещества.
В рабочем журнале отмечают температуру плавления вещества и все изменения, происходящие с ним в процессе нагревания: перемену окраски, разложение и т.п.
Разделение органических жидкостей простой перегонкой
Перегонкой называется процесс, в ходе которого вещество нагревают в соответствующей аппаратуре до кипения и образовавшийся пар конденсируют. Целью перегонки является разделение на компоненты и очистка жидких летучих веществ, имеющих различные температуры кипения (при этом во время процесса наблюдают за температурой). Простую перегонку целесообразно применять для жидкостей с температурой кипения от 40 до 150 °С, загрязненных небольшими количествами примесей, имеющих ничтожное давление пара при температуре кипения очищаемой жидкости, или когда разница в температурах кипения веществ, входящих в состав разделяемой смеси значительна (не менее 80-100 °С).
Если перегоняемая жидкость кипит не выше 120- 130 °С, то применяют проточное водяное охлаждение. При перегонке жидкостей, кипящих выше этой температуры, применяют воздушный холодильник.
При перегонке индивидуальных веществ их температура кипения остается постоянной в течение всей перегонки. Если перегоняется смесь двух веществ, температуры которых различаются значительно, то вначале отгоняется жидкость, имеющая более низкую температуру кипения. Если же температура кипения начинает возрастать, то это означает, что начинает отгоняться другая жидкость, имеющая более высокую температуру кипения, чем первая. В процессе перегонки второй жидкости также устанавливается постоянная температура. Таким образом, меняя приемники, можно собрать несколько фракций, в первых будет преобладать низкокипящая часть перегоняемой смеси, а в последних - высококипящая.
Если перегоняемая смесь состоит из компонентов, температуры кипения которых близки и которые не образуют азеотропных смесей, то применяют дробную или фракционную перегонку. Для этого обычно используют дефлегматоры или ректификационные колонки.
Для веществ, разлагающихся до- или при температуре кипения при атмосферном давлении или имеющих высокую температуру кипения, применяют перегонку при пониженном давлении.
Схема установки для простой перегонки показана на рис.1. Установка состоит из круглодонной колбы 1, дефлегматора 2, термометра 3, нисходящего холодильника Либиха 4, алонжа 5 и приемника 6. Ртутный шарик термометра должен находиться примерно на 0,5 см ниже отводной трубки насадки.
Перед заполнением прибора измеряют объем или вес жидкости, предназначенной для простой перегонки. Заливают жидкость в колбу 1 не более чем на 2/3 её объема. Для равномерного кипения в колбу помещают "кипелки". Включают воду для охлаждения. В качестве теплоносителя используют “баню”, соответствующую температуре кипения наиболее высококипящего компонента перегоняемой смеси. В качестве нагревательного прибора используют электроплитки только с закрытой спиралью. Баню нагревают до температуры, при которой вещество перегоняется с нормальной скоростью (из холодильника поступает в приемник 30-40 капель конденсата в минуту). Контроль за температурой в бане осуществляют с помощью термометра, помещённого в неё. Разность температуры бани и температуры кипения определяется рядом факторов: летучестью отгоняемого вещества, его количеством, конструкцией установки (наличие дефлегматора, высота горла колбы и насадки).
Температура бани, как правило, превышает температуру кипения перегоняемого вещества на 20- 30 °С.
Очистка органических жидкостей перегонкой с водяным паром
Сущность очистки органических жидкостей перегонкой с водяным паром заключается в том, что высококипящие жидкости, не смешивающиеся или мало смешивающиеся с водой, улетучиваются вместе с водяным паром при пропускании его через эти жидкости, затем они вместе с паром конденсируются в холодильнике.
Прибор, используемый при перегонке с водяным паром, изображен на рис. 2. Пар образуется в паровике 1 (вместо него пригодна и колба). Предохранительная трубка 2 служит для выравнивания давления. Паровик заполняют водой приблизительно на половину.

Рис. 2. Прибор для перегонки с паром
Пар через проводящую трубку 4 входит в перегонную колбу 5, в которой находится разделяемая смесь. Обычно эту колбу нагревают. Дистиллят через трубку 6 или насадку Вюрца с термометром поступает в холодильник 7, конденсируется и через алонж 8 стекает в приемник 9.
На тройник 3 надевают короткую резиновую трубку с зажимом. Зажим остается открытым до начала перегонки. В колбу помещают вещество, собирают прибор и подогревают парообразователь, предварительно поместив в него "кипелки". Одновременно подогревают колбу. Как только начнет образовываться пар, резиновую трубку, надетую на тройник, закрывают зажимом.
Спустя некоторое время в приемнике собирается эмульсия, расслаивающаяся при стоянии. Перегонку заканчивают, когда в холодильнике будут образовываться капли чистого дистиллята (воды). Затем открывают зажим на тройнике (если таковой отсутствует, то просто вынимают пароподводящую трубку 4 из перегонной колбы) и выключают парообразователь. Получающиеся в приемнике два слоя: воду и органическое вещество - отделяют друг от друга в делительной воронке, сушат над соответствующим осушителем. В качестве осушителей используют, как правило, безводные неорганические соли. Образующие с водой кристаллогидраты (сульфат натрия, хлорид кальция, перхлорат магния и др.).
Небольшие количества вещества можно перегонять, не пользуясь паровиком, а добавляя некоторое количество воды в перегонную колбу.
ЭКСТРАКЦИЯ. Для работы необходимы: делительная воронка на 100 мл, мерный цилиндр на 50 мл, четыре колбочки по 100 мл (рис. 1), колба на 250-300 мл, бюретка на 50 мл, пипетка на 20 мл, 0,4-0,5 моль/л раствор уксусной кислоты в изоамиловом спирте, 0,1н раствор щелочи, фенолфталеин.
Перед проведением опытов необходимо при помощи воды проверить на герметичность кран и стеклянную пробку делительной воронки. Изоамиловый спирт в количестве 40 мл с растворенной уксусной кислотой вливают в делительную воронку, добавляют 40 мл воды, насыщенной изоамиловым спиртом и проводят экстракцию.
Воду, насыщенную изоамиловым спиртом, получают, смешивая воду и изоамиловый спирт без уксусной кислоты и проводя все нижеописанные операции 1-3. Экстрагирование проводят 4 – 5 раз. Водный слой после каждого экстрагирования объединяют с предыдущими порциями, сливая его в колбу на 250-300 мл.
Последовательность операций при выполнении экстракции
1.Закрыв делительную воронку стеклянной пришлифованной пробкой, правой рукой берутся за горлышко с пробкой, а левой – за кран так, чтобы суженная часть конуса помещалась в ладони, а пальцем можно было бы свободно поворачивать кран. Если держать в ладони сам корпус делительной воронки, то тепло руки повысит давление паров растворителя в воронке, в результате чего пробка и кран могут выскочить.
2.Делительную воронку поворачивают сливной трубкой кверху и осторожно приоткрывают кран. После сброса избыточного давления дают возможность жидкости, которая увлекается струей паров в сливную трубку, стечь обратно в воронку. Закрыв кран, воронку несколько раз встряхивают и снова открывают кран. Интенсивное встряхивание и выравнивание давления в делительной воронке с атмосферным давлением повторяют несколько раз для обеспечения достижения равновесия фаз.
3.Укрепив делительную воронку на штативе, ожидают разделения фаз. Открывают пробку и сливают нижнюю (водную) фазу в колбочку. Верхнюю фазу оставляют в воронке.
Затем в делительную воронку добавляют тот же (40 мл) объем воды, насыщенной изоамиловым спиртом, снова повторяют описанные выше операции 1-3. Экстрагирование проводят 4 – 5 раз. Из слитой каждый раз водной фазы берут пробы по 20 мл и титруют 0,1н раствором щелочи в присутствии фенолфталеина.
Результаты титрования записывают в табл. 1. По формуле (6) вычисляют К
и делают выводы: остается ли К
постоянным, независимым от концентрации уксусной кислоты; если нет, то почему. Сравнивают полученное значение константы распределения с литературными значениями. Обязательно указывают библиографические данные литературного источника.
Сщелочи
=  = =  = =
Таблица
i,
номер экстрагирования
|
Объем, мл |

|
К
|
| Аликвоты водной фазы |
щелочи |
| 1. |
10 Общие правила и порядок работы в химической лаборатории. Правила техники безопасности
Работа в химической лаборатории связана с некоторой опасностью, поскольку многие вещества в той или иной степени ядовиты, огнеопасны и взрывоопасны. Опыт показывает, что большинство несчастных случаев, происходящих в лаборатории, является следствием небрежности и невнимательности работающих.
Возможность несчастных случаев может быть исключена при выполнении всех мер предосторожности. Обычно характер предупредительных мер, обеспечивающих безопасность проведения эксперимента, зависит от вида работы. Однако существуют общие правила, выполнение которых обязательно для каждого работающего в лаборатории, независимо от того, какой эксперимент он проводит.
Работать одному в лаборатории категорически запрещается, так как в ситуации несчастного случая некому будет оказать помощь пострадавшему и ликвидировать последствия аварии.
Во время работы в лаборатории необходимо соблюдать чистоту, тишину, порядок и правила техники безопасности, так как поспешность и небрежность часто приводят к несчастным случаям с тяжелыми последствиями.
Каждый работающий должен знать, где находятся в лаборатории средства противопожарной защиты и аптечка, содержащая все необходимое для оказания первой помощи.
Категорически запрещается в лаборатории курить, принимать пищу, пить воду.
Нельзя приступать к работе, пока учащиеся не усвоят всей техники ее выполнения.
Опыты нужно проводить только в чистой химической посуде. После окончания эксперимента посуду сразу же следует мыть.
В процессе работы необходимо соблюдать чистоту и аккуратность, следить, чтобы вещества не попадали на кожу лица и рук, так как многие вещества вызывают раздражение кожи и слизистых оболочек.
Никакие вещества в лаборатории нельзя пробовать на вкус. Нюхать вещества можно, лишь осторожно направляя на себя пары или газы легким движением руки, а не наклоняясь к сосуду и не вдыхая полной грудью.
На любой посуде, где хранятся реактивы, должны быть этикетки с указанием названия веществ.
Сосуды с веществами или растворами необходимо брать одной рукой за горлышко, а другой снизу поддерживать за дно.
Категорически запрещается затягивать ртом в пипетки органически вещества и их растворы.
Во время нагревания жидких и твердых веществ в пробирках и колбах нельзя направлять их отверстия на себя и соседей. Нельзя также заглядывать сверху в открыто нагреваемые сосуды во избежание возможного поражения при выбросе горячей массы.
После окончания работы необходимо выключить газ, воду, электроэнергию.
Категорически запрещается выливать в раковины концентрированные растворы кислот и щелочей, а также различные органические растворители, сильно пахнущие и огнеопасные вещества. Все эти отходы нужно сливать в специальные бутыли.
В каждой лаборатории обязательно должны быть защитные маски, очки.
В каждом помещении лаборатории необходимо иметь средства противопожарной защиты: ящик с просеянным песком и совком для него, противопожарное одеяло (асбестовое или толстое войлочное), заряженные огнетушители.
В доступном месте в классе-лаборатории должен быть "Уголок техники безопасности", где необходимо разместить конкретные инструкции по методам безопасности работы и правила поведения в химическом кабинете.
При работе в лаборатории необходимо применять индивидуальные средства защиты, а также соблюдать правила личной гигиены.
Контрольная работа №3. Вариант №1
1 На примере
D
- галактозы покажите явление оксо - гидрокситаутомерии Из галактозы получите этил-α-
D
-галактопиранозид. В какой среде данное соединение гидролизуется? Продукты гидролиза назовите
Схема таутомерии выглядит следующим образом:

Получение этил-α-D-галактопиранозида

Как полные ацетали галактозиды гидролизуются в условиях кислотного катализа и устойчивы в разбавленных растворах щелочей. Механизм кислотного гидролиза включает протонирование галактозидного кислорода, расщепление галактозидной С-О связи с образованием галактозил-катиона, который затем атакуется молекулой воды. В результате образуется α-D-галактоза и этанол.
2 Приведите схемы полного метилирования и ацетилирования
[
β
-
D
ксилофуранозы и последующего гидролиза полученных продуктов в присутствии кислоты. Продукты реакций назовите
Метилирование и последующий гидролиз. Продукт метилирования – метил-β -Dксилофураноза, продукт гидролиза - β -Dксилофураноза и метанол

Ацетилирование (продукт – ацетат β -Dксилофуранозы) и последующий гидролиз (продукт – β -Dксилофураноза и уксусная кислота)

1. Напишите схемы реакций лактозы со следующими реагентами: а). уксусным ангидридом; б) гидроксиламином; в) фенилгидразином; г) диметилсульфатом в щелочной среде. Продукты назовите. Укажите значение данных реакций.
а). Реакция с уксусным ангидридом

Продукт – 2,3,4,6,1`,2`,3`6`- октаацетат лактозы
б). реакция с гидроксиламином

Продукт: Оксим лактозы
в). Реакция с фенилгидразином

Продукт – фенилгидразон целлюлозы
г). Реакция с диметилсульфатом в щелочной среде

Продукт – метил - 2,3,4,6,1`,2`,3`6`-окта- О-метиллактоза
2 Напишите структурную формулу дисахарида, состоящего из
p
-
D
-глюку роновой кислоты и 2-
N
-ацетил-4-сульфо-
D
-галактозамина, связанных β-
(
I
→
3) гликозидной связью. В состав какого биополимера входит этот дисахарид?
Структурная формула:
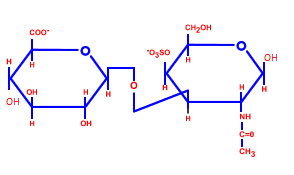
Данный дисахарид является мономером хондроитина, входящего в состав хрящевых тканей
3 Синтезируйте дипептид: валин - лейцин с использованием «защиты» (бензиловым эфиром хлормуравьиной кислоты) и «активации» (этилхлорформиатом). Укажите пептидную связь, С- и
N
-конец, рН среды полученного дипептида


Красным эллипсом обведена пептидная связь, С- и N-концы обведены, соответственно, зеленым и фиолетовым прямоугольниками. pH среды меньше 7.
4 Напишите реакции изолейцина с формальдегидом,
NaOH
,
HCI
,
С2
Н5
ОН (НС1), 2,4-динитрофторобензолом, нингидрином, декарбоксилирования, дезаминирования. Продукты назовите
Реакция с формальдегидом:

Продукт – 2,2-N,N-диметил-3-метилпентановая кислота
Реакция с гидроксидом натрия

Продукт: 2-амино-3-метилвалерат натрия
Реакция с HCl

Реакция с этанолом:

Продукт: этиловый эфир изолейцина.
Реакция с 2,4-динитрофторобензолом:

Продукт: ДНФ-производное изолейцина
Реакция с нингидрином:

Продукты: сине-фиолетовый продукт реакции, вода, углекислый газ, 2-амино-3-метилпентаналь
Реакция декарбоксилирования

Продукт: 1-амино-2-метилбутан
Реакция дезаминирования

Продукт: 3-метилпентен-2-овая кислота
5 Определите строение соединения
C
7
H
7
O
2
N
,
которое широко используется для производства различных красителей и обладает следующими свойствами: а) горит коптящим пламенем; б) растворяется в водном растворе
NaHCO
3
с выделением С
O
2
; в) растворяется в
HCI
;
г) при бромировании бромной водой превращается в дибромопроизводное; д) диазотируется; е) способно к образованию внутримолекулярной водородной связи
Структурная формула соединения (2-аминобензойная кислота):

а). Горение

б).Растворение в водном растворе соды

в). Растворение в соляной кислоте

г).бромирование бромной водой

д). Диазотирование

е) образование водородной связи (отмечена волнистой линией)

6 Осуществите превращения, укажите условия реакций:

Цепочка реакций представлена ниже

7 Напишите строение участка РНК-АУ (Аденин-Урацил). Укажите сложноэфирные и гликозидные связи
    
Синим обозначены сложноэфирные связи, красным – гликозидные.
8 Напишите схемы кислотного и щелочного гидролиза 5' - дезоксиадениловой кислоты. Продукты назовите
Кислотный гидролиз

Продукты: рибофосфат, аденин
Щелочной гидролиз:

Продукты: о-фосфорная кислота, аденозин
Контрольная работа №4. Вариант №1
1
В
чем заключаются особенности электронного строения пятичленных ароматических гетероциклов? Оцените связанную с этим активность пиррола, фурана и тиофена в реакциях электрофильного замещения (в сравнении с бензолом). Приведите примеры реакций
Характерной особенностью пятичленных гетероциклических соединений является одновременное сочетание у них свойств как ароматического соединения, так и диена. Склонность к реакциям того и другого типов, однако, у них различна и связана с природой гетероатома. Так, “ароматические” свойства убывают в ряду: тиофен > пиррол > фуран. При этом их ароматические системы менее устойчивы, чем у бензола.
При нахождении гетероатома в кольце он взаимодействует с его электронной системой по двум направлениям. Как более электроотрицательные элементы, азот, сера и кислород, оттягивают электронную плотность с кольца по индуктивному эффекту, распространяющемуся по системе s- связей. Однако решающий вклад вносит мезомерный эффект, имеющий в каждом из этих случаев противоположное индуктивному эффекту направление. Таким образом, молекула пятичленного гетероциклического соединения становится поляризована, где “положительным” центром поляризации служит гетероатом. Электрические моменты диполей убывают в том же порядке, что и ароматические свойства. Наиболее электроотрицательный кислород имеет меньшую склонность к обобществлению своей пары электронов в ароматической системе, поэтому фуран обладает наименьшими ароматическими свойствами в ряду тиофен-пиррол-фуран.
Меньшая устойчивость ароматических систем у пятичленных гетероциклов объясняется двойственной природой np-электронной пары гетероатома, несоответствием валентных углов внутри цикла значению 120 градусов, характерному для sp2-гибридизованного атома углерода, а также сильной поляризацией связи углерод-гетероатом. В результате наибольшая электронная плотность сосредоточена на ближайших к гетероатому атомах углерода (a- положения). На удаленных от него b- атомах углерода электронная плотность ниже. Все это предопределяет химические свойства соединений этого класса. Пятичленные гетероциклы в целом легче вступают в реакции электрофильного замещения, по сравнению с незамещенным бензолом. Замещение проходит по положению 2, если оно занято, замещаются атомы у третьего атома углерода
Наименее ароматичный фуран, присоединяя в кислой среде протон по атому кислорода, образует диеновую систему, склонную к полимеризции и осмолению. Поэтому реакции электрофильного замещения в фуране (проходящие настолько же легко, как и в фенолах) проводят в нейтральных и щелочных средах. Так, фуран ацилируется ангидридами кислот в присутствии SnCl4
, сульфируется пиридинсульфотриоксидом (мягкий сульфирующий агент), нитруется ацетилнитратом :

Галогенирование фурана галогенами приводит к замещению всех четырех атомов водорода:

Моногалоидные производные получают косвенным путем:

Фуран легко вступает в реакцию Дильса-Альдера с диенофилами (малеиновый ангидрид):

При нагревании с разбавленной соляной кислотой цикл легко раскрывается:

Фурановый цикл приобретает устойчивость при наличии в нем электроноакцепторных заместителей: -NO2
, -CHO, -COOH, -SO2
OH, галогены.
Из производных фурана большое значение имеет применяемый в качестве растворителя тетрагидрофуран, получаемый при гидрировании фурана на никелевом катализаторе.
Тиофен по ароматичности наиболее близок к бензолу и для него характерны все реакции электрофильного замещения, протекающие с большей легкостью, чем у незамещенного бензола. Так, одним из способов очистки технического бензола от тиофена является обработка бензола серной кислотой на холоду:

Образующаяся при этом сульфокислота тиофена растворяется в серной кислоте. Тиофен устойчив в сильнокислых средах, но атом серы чувствителен к окислению, поэтому при нитровании тиофена не применяют азотную кислоту, а используют ацетилнитрат (см. фуран).
При галогенировании в тиофене замещаются только 2 атома водорода:

Бромтиофен легко образует магнийорганические соединения, из которых можно получить многие производные тиофена. При восстановлении тиофена получают тетрагидротиофен (тиофан) (I) , последний может быть окислен в сульфоксид (II) или сульфолан (III):

Вследствие наличия значительной доли положительного заряда у атома азота пиррол в большей степени проявляет кислотные свойства, нежели основные. Тем не менее, это все же очень слабая кислота, способная отдавать протон лишь при взаимодействии с очень сильными основаниями:

Отрицательный заряд аниона (I) значительно делокализован, поэтому в реакциях с галоидными алкилами можно получить как N-замещенные алкилпирролы (при низких температурах), так и a- алкилпирролы, при повышенной температуре):

Свободный пиррол в отличие от тиофена мало устойчив в кислых средах, также проявляя склонность к полимеризации и окислению. Однако, повышенная электронная плотность в кольце приводит и к большей легкости протекания реакций электрофильного замещения, которые проходят в мягких условиях, подобно фурану.
Пиррол имеет сравнительно высокую температуру кипения (130°С), которая объясняется структурированием при образовании межмолекулярных водородных связей:
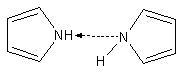
2 На примере пиразола покажите строение пиррольного и пиридинового атомов азота. С помощью химических реакций докажите амфотерный характер пиразола
Строение пиразола:

Атом азота в положении 1 (пиррольный атом) sp3-гибридизован, имеет неподеленную электронную пару и способен участвовать в образовании сопряженной ароматической системы, вследствие чего на этом атоме, при отрыве протона, возникающий отрицательный заряд распределяется по всей системе. Пиридиновый атом азота в положении 2 имеет sp2-гибридизацию и неподеленную электронную пару, которая не участвует в образовании ароматической системы (ее образует двойная связь), что обуславливает его основность.
Реакция с основанием:

Реакция с кислотой:

3 Получите синтезом Скраупа хинолин. Покажите, какое соединение получится, если синтез Скраупа проводить не с акриловым альдегидом, а с бутен
-2
-алем. Напишите для хинолина схемы реакций с НС1, СН3
I
, СН3
С(
O
)С
l
, полученные продукты назовите
Синтез Скраупа представлен ниже:




Аналогичная реакция, только с участием бутен-2-аля:

Реакции хинолина с:
а) HCl

Продукт: N-хлорид хинолина
б) CH3
I

Реакции не происходит
в) CH3
C(O)Cl

Реакции не происходит
4 Осуществите превращения, все продукты назовите

Синтез пиррола

Реакция А

Продукт: пиррол-натрий
Реакция Б.

Продукт: N-метилпиррол
Реакция В

Продукт: 2-нитропиррол
Реакция Г.

Продукт: пирролидин
Реакция Д.

Продукт: N-нитрозопирролидин
5 Напишите структурные формулу тиамина (витамина В1
), назовите гетероциклические системы, входящие в его структуру. Укажите значение тиамина
Структурная формула тиамина:

В его структуру входят такие гетероциклические системы, как пиримидил и тиазол.
Физиологическое значение
В природе тиамин синтезируется растениями и многими микроорганизмами. Животные и человек не могут синтезировать тиамин и получают его вместе с пищей. В тиамине нуждаются все животные за исключением жвачных.
Всасываясь из кишечника, тиамин фосфорилируется и превращается в тиаминпирофосфат.
Тиаминпирофосфат (ТПФ) — активная форма тиамина — является коферментом пируватдекабоксилазного и α-кетоглутаратдекарбоксилазного комплексов, а также транскетолазы. Первые два фермента участвуют в метаболизме углеводов, транскетолаза функционирует в пентозофосфатном пути, участвуя в переносе гликоальдегидного радикала между кето- и альдосахарами. ТПФ синтезируется ферментом тиаминпирофосфокиназой, главным образом в печени и в ткани мозга. Реакция требует присутствия свободного тиамина, ионов Mg2+
и АТФ. Также ТПФ выступает коферментом дегидрогеназы γ-оксиглутаровой кислоты и пируватдекарбоксилазы клеток дрожжей.
Другими производными тиамина являются:
Тиаминтрифосфат, обнаружен у бактерий, грибов, растений и животных, у E. coli играет роль сигнальной молекулы при ответе на аминокислотное голодание.
Аденозинтиаминдифосфат — накапливается у E.coli в результате углеродного голодания.
Аденозинтиаминтрифосфат — присутствует в небольших количествах в печени позвоночных, функция его неизвестна.
6 Напишите схему получения 2,6-дигидроксипурина (ксантина) из мочевой кислоты. Покажите таутомерные превращения ксантина
Получение ксантина из мочевой кислоты:

Схема таутомерных превращений:

7 Сравните основность атомов азота в молекуле алкалоида никотина

Количественно основность можно оценить, исходя из константы основности. Для пиридинового атома азота ее величина составляет Kb
≈ 10-9
, а для алифатического азота Kb ≈ 10-4
. Поэтому пиридиновый атом углерода проявляет основные свойства гораздо в меньшей степени, чем алифатический.
8 Соединение
C
6
H
7
N
даёт соли с кислотами, при действии СН3
I
даёт вещество состава
C
7
H
10
NI
.
При окислении исходное вещество превращается в бета-пиридинкарбоновую кислоту. Какое строение имеет соединение
C
6
H
7
N
?
Напишите указанные реакции
Строение соединения:
9 Напишите структуру триацилглицерина, содержащего остатки олеиновой, линолевой и пальмитиновой кислот, дайте систематическое название. Напишите уравнения кислотного и щелочного гидролиза полученного липида
Соединение носит название 1-олеоил-2-линоил-3-пальмитоилглицерин. Его структура представлена ниже

Кислотный гидролиз

Щелочной гидролиз

10 Напишите структуру лецитина, включающего остатки пальмитиновой и линолевой кислот. Приведите формулу внутренней соли лецитина
Структура данного лецитина следующая:

Формула внутренней соли лецитина

11 Приведите схему синтеза камфоры из пинена. Укажите значение камфоры. Напишите уравнения реакций, доказывающих наличие в молекуле камфоры кетонной группы. Выделите в молекуле камфоры изопреновые звенья
Схема синтеза камфоры из альфа-пинена

Значение камфоры
Камфору используют в медицине и ароматерапии.
В начале XX века камфора широко применялась в производстве некоторых пластмасс, особенно целлулоида — как пластификатор нитрата и ацетата целлюлозы, а также как флегматизатор бездымного пороха.
С давних пор камфору и камфорное эфирное масло применяли для борьбы с молью, но в настоящее время, из-за достаточно высокой токсичности, такое её использование заметно снизилось.
В Китае и по всей юго-восточной Азии, камфору использовали в качестве благовонного курения, для чего кристаллы камфоры помещали на раскаленные угли в курильнице.
В некоторых районах Индии, в основном Бенгалии и Ориссе, натуральную камфору используют в кулинарии, как специю при приготовлении сладостей и молочных пудингов.
Натуральную камфору, желательно борнеосскую, используют при создании химического барометра -штормгласса Роберта Фитцроя.
Вследствие высокой криоскопической константы камфора используется для определения молекулярной массы методом криоскопии.
Применение в медицине
Фармакологи рассматривают камфору, как важный представитель аналептических средств. При введении под кожу растворы камфоры в растительном масле тонизируют дыхательный центр, стимулируют сосудодвигательный центр. Оказывает также непосредственное действие на сердечную мышцу, усиливая обменные процессы в ней и повышая её чувствительность к влиянию симпатических нервов. Под влиянием камфоры суживаются периферические кровеносные сосуды. Способствует отделению мокроты. Возможно, что камфора ингибирует агрегацию тромбоцитов, в связи с чем она рекомендована к применению для улучшения микроциркуляции. Противозудное действие камфоры, возможно, связано с тем, что она, как и ментол, избирательно активирует холодовые рецепторы.
Применяют правовращающую натуральную камфору, добываемую из камфорного дерева, или синтетическую, левовращающую, получаемую из пихтового масла, либо рацемическую.
Кроме того, эфирное масло камфорного лавра применяется в ароматерапии (но только «белое» эфирное масло) — одна из трех фракций исходного масла.
В начале XX века считали, что терапевтическое действие оказывает только натуральная (правовращающая) камфора, затем было доказано, что синтетические продукты — левовращающая и рацемическая камфора не имеют существенного различия с действием натуральной формы. Камфора улучшает альвеолярную вентиляцию, лёгочный кровоток и функцию миокарда. В сборнике рассказов М.А.Булгакова "Записки юного врача" часто всплывает камфора, как что-то первостепенное, необходимое в аптечке врача - действие происходит в 1917 году...
Применяют растворы камфоры в комплексной терапии при острой и хронической сердечной недостаточности, коллапсе, при угнетении дыхания; пневмонии и других инфекционных заболеваниях, при отравлениях снотворными и наркотическими средствами в качестве антидота.
Из производных камфоры в медицине в качестве седативного средства применяется 3-бромкамфора.
Применение камфоры противопоказано при эпилепсии и склонности к судорожным реакциям.
Реакции кетогруппы камфоры


Красными и зелеными линиями обозначены изопреновые звенья

12 Осуществите превращение:

Укажите значение витамина Д2
.
Синтез витамина Д2
(эргокальциферола)

Значение витамина Д2
Эргокальциферо́л (витамин Д2
) — вещество, регулирующее обмен кальция и фосфора. Если витамина D не хватает, то из организма выводится большое количество солей кальция и фосфора. Костная ткань, которая является почти единственным местом их накопления, начинает быстро терять эти элементы. При этом кости становятся мягкими, искривляются и легко ломаются.
Человек получает витамин D двумя путями: с пищей и из собственной кожи, где он образуется под действием ультрафиолетовых лучей. Вот почему у детей, растущих при недостатке солнечного света, развивается рахит.
Эргокальциферол очень токсичен, доза 25 мг уже опасна (20 мл в масле). Плохо выводится из организма, что приводит к кумулятивному эффекту. Основные симптомы отравления тошнота, обезвоживание, гипотрофия, вялость, повышение температуры тела, мышечная гипотония, сонливость, сменяющаяся резким беспокойством, судорогами.
|