«Тонкослойная хроматография остаточных концентраций пестицидов в пищевых продуктах»
Содержание
Введение
Глава 1.Основы планарной (тонкослойной) хроматографии
Глава 2. Состояние и перспективы использования современных инструментальных методов анализа пестицидов
Глава 3. Методические указания по определению хлорорганических пестицидов в воде, продуктах питания, кормах и табачных изделиях хроматографией в тонком слое
Глава 4. Современное аппаратурное оформление
Литература
Введение
Химикаты (инсектициды, гербициды, фунгициды) используются для удобрения почвы, борьбы с сорняками, насекомыми и грызунами, для защиты урожая от плесени и грибков. С их помощью повышают урожайность, увеличивают срок хранения растений, улучшают внешний вид фруктов, овощей и зерна. Сегодня предлагается выбор из 5000 видов пестицидов и 700 химических ингредиентов. По сравнению с началом 40-х гг., когда были впервые использованы пестициды, их потребление в сельском хозяйстве возросло в десятки раз, а потери урожая из-за насекомых за последние 50 лет увеличились вдвое. Эта статистика ставит под сомнение "эффективность" пестицидов. Интересно, что применение пестицидов привело к развитию 650 видов вредителей, устойчивых к некоторым из этих ядов.
Каждый день в мире около 3000 человек отравляются пестицидами. Это более миллиона отравлений в год химическими веществами, загрязняющими воздух, почвы, воду и продукты. Отдельно по Европе эти цифры не менее шокирующие. Только в 2005 году страны ЕС начали пытаться ввести единые стандарты в оценке опасности химических веществ, попадающих в продукты питания, и единую маркировку для продуктов питания. Известно, что многие пестициды опасны для здоровья и обладают канцерогенными свойствами, однако до сих пор покупатель не может по этикетке определить, насколько же насыщен покупаемый продукт этими неполезными веществами. В развитых странах у потребителя, в принципе, существует выбор - покупать "органическую" (выращенную без химикатов) продукцию, или обычную. Разница в цене весьма существенна, и выбор "органических" продуктов не столь велик, как обычных.
Организация по защите окружающей среды допускает, что из 320 пестицидов, разрешенных к применению в агрономии, по меньшей мере, 66 -
предполагаемые канцерогены. Многие из этих пестицидов смешиваются с 1200 нейтральными ингредиентами, состав которых производители не обязаны разглашать, ссылаясь на "коммерческую тайну". Для 800 из них до сих пор не установлены уровни токсичности, они предположительно являются канцерогенами, поэтому необходимо использовать методы идентификации пестицидов в продуктах питания.
ГЛАВА 1. ОСНОВЫ ПЛАНАРНОЙ (ТОНКОСЛОЙНОЙ) ХРОМАТОГРАФИИ
Планарная (тонкослойная)хроматография
Тонкослойная (планарная) хроматография занимает одно из ведущих мест в качественном и полуколичественном анализе сложных природных, фармацевтических, медикобиологических и химических объектов. Среди других хроматографических методов планарную хроматографию отличают следующие достоинства и особенности:
- это единственный хроматографический метод, позволяющий проводить полный анализ неизвестной смеси, поскольку исследователь имеет возможность проверить, не остались ли на старте неэлюированные компоненты;
-по производительности превосходит газовую и
высокоэффективную жидкостную хроматографию, по крайней мере, на порядок; использует более простое и дешевое оборудование;
- обладает высокой селективностью, которую легко варьировать, подбирая состав подвижной фазы; в отличие от ВЭЖХ нет ограничений в выборе растворителей;
- дает возможность одновременного разделения нескольких образцов; использования однократного или многократного элюирования (при различных условиях), а также одновременного разделения компонентов одного и того же образца с помощью различных элюентов;
- возможна оптимизация разрешающей способности
хроматографической системы при разделении сложной смеси только для интересующих компонентов, что позволяет экономить время;
- возможно детектирование соединений с высокой
чувствительностью и селективностью, которые легко варьировать подбором проявляющего реагента; полученные результаты разделения легко оценить визуально;
можно сохранять хроматограммы для последующего
детектирования и осуществлять спектральную идентификацию
хроматографических зон после разделения в любом диапазоне длин волн, включая ИК.
у планарной хроматографии есть и некоторые недостатки:
- ограниченная разделяющая способность из-за сравнительно небольшой длины разделяющей зоны (3-10 см);
- чувствительность ниже, чем в случае ВЭЖХ;
- зависимость результатов анализа от окружающей среды: относительной влажности, температуры, а также наличия загрязняющих веществ в воздухе;
- трудности в работе с образцами, имеющими высокую летучесть, а также с веществами, чувствительными к действию кислорода воздуха или света.
Классическая, наиболее простая и широко используемая методика тонкослойной хроматографии включает проведение следующих основных операций:
1)нанесение анализируемой пробы на слой сорбента;
2)разделение компонентов пробы на отдельные зоны в потоке подвижной фазы;
3) обнаружение зон на слое сорбента (часто реагентом, образующим с разделенными веществами окрашенные соединения);
4) количественная оценка полученного разделения, включая определение величины удерживания и определение содержания вещества в зонах на хроматограмме.
Положение зоны вещества на хроматограмме характеризуется величиной Rf
, которая равна отношению расстояния от стартовой линии до центра зоны вещества к расстоянию от стартовой линии до линии фронта. Значение Rf
- величина постоянная для данного соединения в данной истеме и зависит от ряда условий: способа элюирования, качества и активности сорбента, толщины слоя, качества растворителей, количества нанесенного вещества, длины пробега растворителей, положения стартовой линии и почти не зависит от температуры. По этой величине проводят идентификацию компонентов в смеси.
На качество разделения компонентов смеси в планарной хроматографии влияет большое число факторов: тип разделительной камеры; предварительное насыщение камеры и слоя сорбента парами подвижной фазы; стартовый размер пятна; расстояние от старта до нижнего края пластинки; относительная влажность воздуха лабораторного помещения; средний диаметр частиц и их форма; толщина и равномерность нанесения слоя сорбента; наличие микроповреждений слоя; тип вещества, связывающего сорбент; скорость элюирования; объем растворителя в камере; наличие примесей в элюенте; конвекция в газовой фазе внутри камеры.
Для разделения смесей веществ в тонком слое сорбента применяют адсорбционную, распределительную и ионообменную хроматографию, отличающиеся, прежде всего характером взаимодействий между растворенными веществами и твердой или жидкой фазами, с которыми они соприкасаются. На практике эти взаимодействия почти никогда не протекают изолированно, и разделение веществ обусловлено несколькими взаимодействиями. При выборе подходящего варианта хроматографии в первую очередь следует обратить внимание на строение разделяемых веществ. При помощи адсорбционной и распределительной хроматографии разделяются вещества, строение которых различается природой, числом и характером полярных и неполярных заместителей. При хроматографировании в тонком слое сорбента чаще всего применяют адсорбционную хроматографию, которая проще по выполнению, более эффективна, а результаты анализа более воспроизводимы.
Сорбенты в тонкослойной хроматографии
В качестве сорбентов в ТСХ применяют материалы, которые отвечают следующим требованиям: образуют химически и физически стабильные слои; не образуют ковалентных связей с разделяемыми веществами; не растворяются в подвижной фазе или перемещаются вместе с ней по пластинке; не содержат компонентов, мешающих разделению или детектированию; не имеют собственной окраски; не набухают и не сжимаются под действием подвижной фазы.
В качестве подложки для сорбента используется стекло, алюминиевая фольга, полимерные пленки (полиэтилентерефталат). Для придания стабильности слоя сорбента на подложке используются различные связующие вещества: гипс (5-10%), силиказоль, силикаты щелочных металлов, полиакриламид, полиакриловый эфир, крахмал. К адсорбенту часто добавляют флуоресцентный индикатор для детектирования веществ, поглощающих в УФ-области спектра. С этой целью используют: смесь силикатов цинка и магния; смесь сульфидов цинка и кадмия; вольфраматы щелочноземельных элементов.
Большое значение, особенно для эффективности разделения, имеют такие характеристики сорбентов, как диаметр частиц, среднее распределение частиц по размерам и размер пор. В классической тонкослойной хроматографии для производства пластинок используются частицы с размером 5 - 20 мкм. Для высокоэффективной тонкослойной хроматографии (ВЭТСХ) необходим сорбент, диаметр частиц которого составляет 5 - 7 мкм. Сравнение характеристик пластинок для ТСХ и ВЭТСХ приведено в таБЛ.22. Монолитные сорбенты представляют собой новое поколение стационарных фаз, которые могут быть использованы и в планарной хроматографии получают прямой сополимеризацией метакриловых полимеров, например, сополимера глицинметакрилата и этилендиметакрилата. Монолитные стационарные фазы не содержат частиц, а роль разделительного пространства выполняют поверхность и объем проточных каналов (пор). Макропористая структура монолитных сорбентов содержит как минимум два вида пор: макро- и мезопоры. Преимущества таких носителей заключаются в заметном повышении скорости и эффективности разделения, так как для них отсутствуют обычные диффузионные ограничения межфазного массообмена.
Таблица 1. Сравнение характеристик пластинок для классической (ТСХ) и высокоэффективной (ВЭТСХ) тонкослойной хроматографии.
| Характеристики |
ТСХ |
ВЭТСХ |
| Средний размер частиц, мкм |
5 -20 |
5-7 |
| Толщина слоя, мкм |
250 |
100,200 |
| Количество проб |
12 |
36 -72 |
| Длина пробега фронта растворителя, мм |
100 - 150 |
30 - 50 |
| Время разделения, мин |
30 - 200 |
3 -20 |
| Количество растворителя, мл |
50 |
5 -10 |
| Предел детектирования, нг |
| поглощение |
100 - 1000 |
10 - 100 |
I |
| I |
| флуоресценция |
1 - 100 |
0,1 - 10 |
I |
Основные типы сорбентов, используемых в ТСХ
Силикагель
полярный адсорбент, содержит активные силанольные и силоксановые группы, его применяют для разделения соединений различной полярности.
Оксид алюминия
полярный адсорбент с гетерогенной поверхностью, содержит активные ОН-группы, обладает заметно выраженными протоноакцепторными свойствами; его применяют для разделения ароматических углеводородов, алкалоидов, хлоруглеводородов, стероидов
Флоросил - основной силикат магния, занимает промежуточное положение между оксидом алюминия и силикагелем; удобен для разделения флаваноидов, стероидов и ацетилированных углеводородов
Полиамиды - группа полярных сорбентов со смешанным
механизмом разделения: карбоксамидная группа ответственна за адсорбционный механизм, метиленовые звенья - за распределительный механизм. Эти сорбенты применяют для разделения пищевых красителей, флаваноидов, танинов, нитрофенолов, спиртов, кислот.
Модифицированные силикагели с привитыми группами (амино, циано, диол-, C2
-,Cg
-, C1g
-), отличными по полярности.
Важной характеристикой сорбента является его активность, она зависит от содержания воды и понижается при увеличении содержания воды в сорбенте.
Для успешного разделения смесей веществ большое значение имеет выбор сорбента. В первую очередь нужно исходить из свойств разделяемых соединений: их растворимости (гидрофильности, гидрофобности), содержания и характера функциональных групп. Насыщенные углеводороды адсорбируются слабо или совсем не адсорбируются на силикагелях и оксиде алюминия. Введение двойных связей, особенно сопряженных, увеличивает адсорбционную способность соединений.
Функциональные группы в еще большей степени усиливают способность веществ к адсорбции. Адсорбционная способность функциональных групп возрастает в следующем порядке:
СН=СН<ОСНз<СООR <C=O<CHO<SH<NН2
<OH<COOH.
Для количественной оценки содержания вещества в хроматографическux зонах используют различные методы:
1. Определение с удалением хроматографической зоны с пластинки можно проводить двояким образом: переносом хроматографической зоны вместе с сорбентом либо экстрагированием хроматографической зоны со слоя сорбента.
2. Определение соединений непосредственно на пластинке методом визуального сравнения размеров площадей пятен и их окраски с соответствующими параметрами пятен стандартных образцов
3. Метод денситометрии, повышающий точность результатов определения, основан на сканировании хроматограмм в видимом и УФсвете с помощью «хроматографических спектрофотометров» денситометров. Денситометры позволяют измерять поглощение света веществом на хроматограмме в режиме пропускания или отражения, а также флуоресценцию и ее тушение. Режим пропускания доступен, если только исследуемое вещество имеет полосу поглощения в видимой области спектра. В У Ф-области регистрацию в режиме про пускания осуществить нельзя из-за собственного поглощения силикагеля и подложки хроматограммы.
4. Метод вuдеоденсuтометрuu - сравнительно новый метод для количественной обработки хроматограмм. Принцип метода заключается во введении изображения хроматограммы в компьютер с помощью видеокамеры или цифровой камеры с последующим сравнением интенсивностей пятен стандартных и определяемых соединений. Видеоденситометр включает осветительный блок, видеокамеру с платой видеоввода или сканер, персональный компьютер с установленной операционной системой Windows и соответствующим программным обеспечением. В России такие комплексы производят НТЦ «Ленхром» (г. С.-Петербург) - денситометр «ДенСкан-О4» и «Сорбполимер» (г. Краснодар) денситометр «Сорбфил». Программа обработки хроматографических данных позволяет выполнять следующие функции: вводить изображения хроматограмм и сохранять их с высоким качеством и разрешением; выделять на введенном изображении хроматограммы рабочий участок, на котором будет производиться дальнейшая обработка изображения; производить
автоматический или ручной поиск пятен; проводить обработку пятен, переводить их в форму хроматографических пиков, рассчитывать значения Rr
и площади пиков; измерять содержание вещества в анализируемых пятнах (в относительных единицах); вводить значения концентраций для построения градуировочных зависимостей: линейной интерполяцией; линейной аппроксимацией более чем, через две точки; квадратичной интерполяцией; автоматически вычислять содержание вещества в анализируемых пятнах по введенным калибровочным значениям; представлять результаты в виде печатных документов. [1-3]
Количественную обработку пятна в видеоденситометрии проводят по двум характеристикам: по площади пятна и его «объему» в пространстве, при этом в качестве третьей координаты используют яркость (интенсивность окраски пятна) (рис. 1).
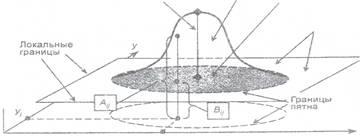
Рис. 1. Вид пространственного распределения яркости в области пятна:
Ai,j - значение уровня яркости точки пятна; Bi,j- значение уровня яркости точки на базовой поверхности.
5. Денсuтометрuя с планшетным сканером с программным обеспечением для обработки хроматограмм практически не отличающимся от стандартных программ, применяемых для видеоденситометров, но существенно меньшей стоимости. При этом сканирование дает более четкое изображение хроматографических зон, что можно объяснить пониженным влиянием неравномерности освещения анализируемых объектов, чем в случае видеоденситометра.
Прuменение для решения практическux задач. Применение тех особенно эффективно для предварительного разделения (по классам, группам, видам веществ) компонентов сложных смесей органических загрязнителей воды, почвы и воздуха. Индивидуальная идентификация с помощью одной лишь тех затруднена из-за отсутствия высокочувствительных и селективных детекторов, кроме того, определение целевых компонентов менее точно, чем в случае ГХ и ВЭЖХ. Часто ТСХ применяют на первом этапе анализа для разделения сложных и многокомпонентных смесей органических соединений на отдельные более простые группы, и уж потом проводят более детальное исследование этих групп «более тонкими» методами (ГХ, ВЭЖХ, ЯМР, ИК или масс-спектрометрией).
Использование ТСХ при анализе загрязненной пресной и морской воды открывает широкие возможности для препаративного разделения, предшествующего другим методам, разделения искомых примесей и дополнительной идентификации. ТСХ используют для обнаружения и
полуколичественного определения веществ разной природы: поверхностно-активных веществ, углеводородов, ПАУ, фенолов, пестицидов.
Для определения неионных ПАВ в сточных и речных водах используют пластинки со слоем силикагеля или Кизельгеля о. На пластинку наносят хлороформенный экстракт ПАВ и разделяют их при использовании в качестве подвижной фазы смесей этилацетат: вода: уксусная кислота. Обнаруживают пятна при опрыскивании смесью: реактив Бургера: фосфорная кислота: этанол 5% раствор BaCI2
.2H2
0 (10:1:10:5). ПАВ проявляют в виде розовых пятен. Метод позволяет определить в воде от 0,1 до 1,0 мг/л неионогенных ПАВ. Из сточных вод в этих условиях экстрагируются ионные ПАВ, но они движутся вместе с фронтом растворителя и не проявляются.
Предложено много методик определения фенолов. Хлорфенолы разделяют на пластинках с оксидом алюминия при многократном элюировании бензолом или на силикагелевых пластинках при элюировании смесью бензола и петролейного эфира (1: 1). Определяют фенолы проявлением 2% раствором 4-аминоантипирина (предел обнаружения 0,5 мкг/л) или по флуоресценции при 254 нм (до 0,5 мкг фенолов). Второй вариант определения фенолов - разделение в виде: антипириновых, 4-аминоантипириновых производных или с п- нитрофенилазокрасителями.[4-6]
ГЛАВА 2. СОСТОЯНИЕ И ПЕРСПЕКТИВЫ ИСПОЛЬЗОВАНИЯ СОВРЕМЕННЫХ ИНСТРУМЕНТАЛЬНЫХ МЕТОДОВ АНАЛИЗА ПЕСТИЦИДОВ В УКРАИНЕ
Увеличение масштабов и ассортимента применения пестицидов в сельскохозяйственной практике продолжает стимулировать разработку и использование методов аналитической химии малых концентраций токсических органических веществ для анализа объектов окружающей среды, сельскохозяйственного сырья, кормов и продуктов питания. Определение остатков пестицидов в этих средах не имеет самостоятельного значения, но является необходимой частью общей информации для достижения адекватной оценки риска, связанного с применением пестицидов. Оценка риска в прошлом была связана главным образом с безопасностью человека, и по этой причине определение остатков пестицидов было сосредоточено, главным образом, на сельскохозяйственном сырье и продуктах питания. В последние годы увеличение внимания к влиянию пестицидов не только на человека, но и на его окружение, требует значительно большей информации по остаточным количествам не только применяемых пестицидов, но и продуктов их разрушения и метаболизма в различных средах. Изучение остатков пестицидов теперь включает все виды сельскохозяйственного сырья, кормов и продуктов питания, воду, воздух и почву. Это в сочетании с внедрением в сельскохозяйственные технологии пестицидных препаратов с низкими нормами расхода (<10 г/га) требует принципиально новых подходов и методов для идентификации и количественного определения остатков пестицидов в различных средах.
Принимая во внимание объем необходимой информации, который должен быть получен в результате анализа различных матриц и сред, методика выполнения измерений (МВИ) остатков пестицидов должна отвечать большинству или всем следующим требованиям:
— обеспечивать достоверное отделение анализируемого вещества от мешающих примесей;
— обеспечивать однозначную идентификацию анализируемого вещества;
— иметь низкий предел количественного определения;
— иметь короткое время анализа;
— иметь низкую стоимость;
— обеспечивать разумную степень точности и правильности результатов;
— обеспечивать надежность получаемых результатов.
Стремление разработчиков методик как можно полнее удовлетворять этим требованиям является одним из основных стимулов в совершенствовании МВИ. Современная МВИ, основанная на инструментальных методах анализа, подразделяется на следующие стадии:
— экстракция анализируемых пестицидов и их метаболитов;
— очистка полученного экстракта;
— возможное получение производных анализируемых пестицидов и продуктов их разрушения и метаболизма;
— хроматографическое разделение
— определение (детектирование) анализируемых веществ.
Способ экстракции, который используется в МВИ, должен обеспечивать количественное и селективное извлечение определяемых веществ, т. е. максимально извлекать из анализируемой матрицы определяемые вещества на фоне как можно меньшего извлечения коэкстрактивных (мешающих) веществ. В противном случае потребуется более сложная стадия очистки полученного экстракта, что неизбежно приведет к потерям определяемых веществ и увеличению общей ошибки анализа. В связи с этим в настоящее время проявляется общая тенденция в анализе остатков пестицидов использовать способы экстракции, которые легко поддаются автоматизации, уменьшают число операций, выполняемых вручную, и количества используемых органических растворителей и обеспечивают возможность анализа большого числа проб. Этим требованиям отвечает твердофазная экстракция (ТФЭ), которая является альтернативой традиционной экстракции в системе жидкость-жидкость и которая позволяет объединить отбор проб с концентрированием. Использование готовых коммерчески доступных патронов (картриджей) для ТФЭ значительно упрощает процедуру подготовки проб к анализу по сравнению с традиционными способами. ТФЭ используется не только в анализе воды, но также в анализе почвы, фруктов, овощей и других пищевых продуктов. Из экстрактов этих матриц, полученных с использованием малополярных и неполярных органических растворителей, пестициды затем концентрируют на молекулярных сорбентах за счет диполь-дипольных взаимодействий или образования водородных связей. Для этих целей используют картриджи, заполненные силикагелем, флоризилом или оксидом алюминия. Нами проведены систематические исследования процесса динамической сорбции следовых количеств пестицидов различных классов на макросетчатом "сверхсшитом" сополимере стирола с дивинилбензолом (полисорб. В результате этих исследований разработан сорбционный способ концентрирования с использованием пластмассовых патронов-концентраторов, заполненных полисорбом, который позволяет проводить экспресс-определение пестицидов в воде на 1-2 порядка ниже значений ПДК. Интересно отметить, что в совместном проекте SMT4-CT96-2142 семи Европейских исследовательских центров Франции, Бельгии, Германии, Нидерландов, Испании и Португалии, который стартовал в 1997 году и предметом которого являлась разработка метода определения множественных остатков пестицидов в питьевой воде с помощью ТФЭ, позволяющего контролировать пестициды в воде на уровне 0,1 мкг/л (в соответствии с требованиями Европейской Директивы по питьевой воде 80/778/EEC), были исследованы девять сорбентов различных фирм на основе С18-обращенной фазы и SDB-1 [10]. В результате этих исследований было установлено, что наиболее подходящим сорбентом для ТФЭ пестицидов из воды оказался SDB-1 — сорбент на основе сополимера стирола с дивинилбензолом, эффективность которого для этих целей была нами установлена еще в начале 80-х годов прошлого столетия.
В последние годы для извлечения пестицидов из различных матриц находит применение сферхкритическая флюидная экстракция (СФЭ), которая рассматривается как альтернатива обычной жидкостной экстракции в аппарате Сокслета. В качестве сверхкритических флюидов используются диоксид углерода, оксид азота и смеси диоксида углерода и оксида азота с метанолом и толуолом. При сверхкритических условиях (температура 40 °С, давление 300 атм) сольватирующие свойства диоксида углерода подобны таковым фреонов или гексана. Одно из основных преимуществ СФЭ заключается в том, что при этом из анализируемых матриц извлекаются остатки различных пестицидов и продуктов их разрушения и метаболизма, которые не экстрагируются традиционными методами, даже при проведении экстракции в аппарате Сокслета. Аппаратурное оформление ТФЭ позволяет полностью автоматизировать этот процесс. Украинским химикам-аналитикам, работающим в области анализа пестицидов, еще только предстоит знакомство с этим мощным средством извлечения остатков пестицидов из почвы, растительного материала и животных тканей, позволяющим проводить экстракцию большого количества проб. Особенно впечатляет эффективность ТФЭ для анализа таких супертоксикантов, как полихлорированные дибензодиоксины и полихлорированные дибензофураны.
В качестве способа очистки экстрактов в анализе остатков пестицидов в настоящее время часто применяется гель-хроматография либо как самостоятельный способ, или как ступень в многостадийной операции очистки. Особенно эффективен этот способ очистки при анализе матриц, содержащих большое количество липидов. Наибольшее использование для этого способа очистки получили гели, работающие в среде органических растворителей. Разработаны автоматизированные установки, позволяющие очищать большое количество проб без какого-либо внимания со стороны персонала лаборатории. Эффективность этого способа очистки была впервые продемонстрирована нами в отечественных исследованиях для очистки экстрактов из риса, содержащих гербициды сатурн и префикс, при использовании гелей, образованных слабосшитыми сополимерами стирола с дивинилбензолом, хорошо набухающими в малополярных и неполярных органических растворителях.
Гель-хроматография является обязательной стадией многоступенчатой операции очистки при разработке и использовании так называемых методик определения множественных остатков (multiresidue) пестицидов. Увеличение числа применяемых пестицидов и источников их поступления в объекты окружающей среды, сельскохозяйственное сырье и продукты питания обусловливает значительное увеличение объема химико-аналитических исследований. Естественно, что использовать для определения каждого пестицида в каждой анализируемой матрице отдельную МВИ экономически невыгодно и неудобно. Значительно более привлекательны такие методические подходы, которые позволяют охватить все количество применяемых в сельскохозяйственной практике пестицидов несколькими МВИ. Такой подход имеет ряд важных преимуществ:во-первых, общее время анализа существенно сокращается; во-вторых, общее число пестицидов и их метаболитов, которые могут быть определены этими методиками, резко увеличивается и, в третьих, эти методики могут быть при необходимости быстро адаптированы к новым анализируемым матрицам и к новым пестицидам. В настоящее время за рубежом для контроля за содержанием пестицидов используются только методики определения множественных остатков пестицидов, которые позволяют проводить определение в одной пробе сельскохозяйственного сырья, пищевого продукта, воды, почвы или воздуха практически всех пестицидов, которые используются в сельскохозяйственной практике. Так, например, методика определения множественных остатков АОАС 990.06 позволяет проводить определение в одной пробе питьевой воды 29 хлорорганических пестицидов. Методика определения множественных остатков АОАС 991.07 предназначена для определения 44 азот- и фосфорорганических пестицидов в одной пробе питьевой воды. Методика определения множественных остатков Министерства здравоохранения Германии S 8 предназначена для определения в одной пробе фруктов или овощей 91 хлор-, фосфор- и триазиновых пестицидов. Методика определения множественных остатков S 19 (Германия) позволяет проводить определение в одной пробе почвы 220 хлор-, фосфор- и азотсодержащих пестицидов. Методика Европейского проекта SMT4-CT96-2142 позволяет определять в одной пробе питьевой воды 38 пестицидов, являющихся приоритетными для стран-разработчиков методики.
К сожалению, в Украине до настоящего времени при разработке МВИ, предназначенных для контроля за содержанием остатков пестицидов, используется подход, который был сформирован в недрах Государственной комиссии по химическим средствам борьбы с вредителями, болезнями растений и сорняками бывшего СССР, и заключающийся в необходимости разработки отдельной методики для каждого пестицида и каждой анализируемой матрицы. В основу такой разработки кладутся методики, которые представляет фирма-разработчик пестицидного препарата вместе с отчетом о валидации представляемых методик независимой лабораторией и результатами полевых испытаний по определению остатков пестицидов в сельскохозяйственных культурах, почве, воде и воздухе рабочей зоны. Эти методики фирма-разработчик пестицидного препарата представляет только для прохождения процедуры государственной регистрации пестицида в Украине для того, чтобы показать, что данные по остаткам пестицида в сельскохозяйственных культурах, почве, воде и воздухе рабочей зоны, которые представляет фирма, получены с помощью валидированных методик. Таким образом, МВИ, представляемые фирмой-разработчиком пестицидного препарата, служат только для целей государственной регистрации пестицида и не являются МВИ, с помощью которых в стране-разработчике пестицидного препарата осуществляется контроль за содержанием остатков пестицидов в сельскохозяйствнном сырье, продуктах питания и объектах окружающей среды. Прерогатива разработки МВИ, предназначенных для контроля за содержанием остатков пестицидов в различных средах, за рубежом закреплена не за фирмами-производителями пестицидных препаратов, а за министерствами и ведомствами, которые ответственны за ту или иную область контроля. Например, в США это Агенство по охране окружающей среды (EPA) и Администрация по контролю пищевых продуктов и лекарственных препаратов (FDA).
Таким образом, для разработки современной стратегии использования МВИ для определения остатков пестицидов в Украине необходимо четко разграничить МВИ, которые необходимы для целей государственной регистрации пестицидов, и МВИ, которые предназначены для государственного санитарно-эпидемиологического надзора за применением пестицидов. Для целей государственной регистрации пестицидов экономически и методически оправдан следующий подход к разработке МВИ: один пестицид — одна сельскохозяйственная культура/среда — одна МВИ. В основу разработки таких МВИ кладутся методики, которые представляют фирмы-разработчики пестицидных препаратов. Разработанные таким образом МВИ используются при определении остатков пестицидов в сельскохозяйственном сырье, почве, воде и воздухе рабочей зоны только при проведении предрегистрационных государственных испытаний пестицидов. Для целей государственного санитарно-эпидемиологического надзора за применением пестицидов конечно же необходимы МВИ, в основу разработки которых положен принцип определения множественных остатков пестицидов в одной пробе. Использование таких МВИ значительно удешевит как их разработку, так и последующее проведение санитарно-эпидемиологического надзора за применением пестицидов. В настоящее время рассматривается вопрос о возобновлении функционирования системы мониторинга пестицидов, которая в свое время (1984-1991 гг.) была разработана во ВНИИГИНТОКС (теперь Институт экогигиены и токсикологии им. Л.И.Медведя) и внедрена в практику работы сети санэпидстанций МЗ Украины. В основе такого мониторинга должны лежать только методики определения множественных остатков пестицидов. Нами проанализированы химико-аналитические аспекты функционирования в прошлом унифицированной системы контроля за остаточными количествами пестицидов в сельскохозяйственном сырье, продуктах питания и объектах окружащей среды, намечены пути для модернизации этой системы и методические подходы для разработки методик определения множественных остатков пестицидов в фруктах, овощах и воде.
Хроматографические методы продолжают оставаться основным инструментом аналитической химии пестицидов. По темпам развития среди них первые места занимают капиллярная газовая хроматография (ГХ), высокоэффективная жидкостная хроматография (ВЭЖХ) и хромато-масс-спектрометрия (ГХ/МС, ЖХ/МС). Капиллярная ГХ не имеет альтернативы при разработке методик определения множественных остатков пестицидов.
Ряд пестицидов, используемых в сельском хозяйстве Украины, не может быть подвергнут непосредственному газохроматографичекому определению вследствие их низкой летучести или недостаточной термической стабильности. Для того, чтобы сделать возможным определение этих соединений с помощью ГХ их превращают в различные производные. Такая операция обычно повышает летучесть и уменьшает адсорбцию хроматографируемых соединений на твердых носителях, увеличивает их термостойкость и улучшает разделение. В некоторых случаях при этом достигается также и значительное увеличение чувствительности детектирования полученных производных. Все это является предметом реакционной газовой хроматографии. Нами впервые в отечественных исследованиях была показана эффективность использования реакционной газовой хроматографии в анализе пестицидов на примере определения остаточных количеств гербицидов — производных феноксиалканкарбоновых кислот (2,4-Д, 2,4-ДМ) в продуктах питания. С тех пор метод реакционной газовой хроматрографии широко используется в лабораториях Института при проведении государственных испытаний пестицидов и осуществлении государственной санитарно-гигиенической экспертизы.
Метод ВЭЖХ продемонстрировал определенные преимущества при совместном определении пестицидов и их метаболитов в одной пробе. Это в особой степени касается тех пестицидов, которые невозможно определять с помощью ГХ вследствие их термической нестабильности, высокой полярности и низкой летучести. Использование ВЭЖХ в анализе пестицидов позволяет обойтись без трудоемкой операции получения производных. Институт одним из первых в Украине начал использование этого метода для определения пестицидов. В настоящее время ВЭЖХ — рутинный метод анализа во многих лабораториях Института. Особенно широко этот метод используется при проведении государственной санитарно-гигиенической экспертизы пищевых продуктов.
Перечисляя хроматографические методы, которые используются в анализе остатков пестицидов, нельзя не упомянуть и метод тонкослойной хроматографии (ТСХ), который был открыт в 1938 г. украинскими учеными Н.А.Измайловым и М.С.Шрайбер. Полуколичественный вариант ТСХ является и в настоящее время недорогим и эффективным методом разделения, идентификации и полуколичественного определения остатков пестицидов. Именно полуколичественный вариант ТСХ сыграл большую роль в становлении химико-аналитической службы Министерства здравоохранения Украины для контроля за содержанием остатков пестицидов в продуктах питания и объектах окружающей среды, когда методы ГХ и ВЭЖХ еще не были доступны для широкого использования. Во многом это произошло благодаря работам, выполненным в стенах Института. В настоящее время ТСХ в анализе остатков пестицидов в основном используется как альтернативный аналитический метод для подтверждения правильности идентификации пестицидов, полученной с помощью методов ГХ и ВЭЖХ. ТСХ незаменимый инструмент и в анализе остатков пестицидов, когда требуется проверить очень большое число проб пищевых продуктов или объектов окружающей среды на наличие пестицидов. В таких случаях обычно применяется методология скрининга. Все пробы, давшие "положительную" реакцию, далее исследуют каким-то более специфическим инструментальным методом (ГХ, ВЭЖХ, ГХ/МС, ЖХ/МС), в то время как все отрицательные результаты скрининга принимают как окончательные без какой-либо проверки. Институт распологает комплектом оборудования для количественной ТСХ (фирма КАМАГ, Германия). Тем не менее перспективы дальнейшего использования ТСХ в анализе пестицидов прежде всего следует связывать с полуколичественным вариантом этого метода. Альтернативы этому нет.
Каждый этап применения пестицидов в мировой сельскохозяйственной практике с конца 40-х годов прошлого столетия и до настоящего времени может быть охарактеризован своими собственными химико-аналитичесчкими проблемами. Однако одна проблема в анализе остатков пестицидов остается неизменной — необходимость постоянного снижения пределов количественного определения (limit of quantitafication, LOQ) пестицидов. Достижение очень низких пределов количественного определения при использовании МВИ сопровождается уменьшением уровня достоверности (надежности идентификации) результата анализа. Часто для того, чтобы достичь очень низких пределов количественного определения необходимо использовать сложную многостадийную процедуру очистки и стадию получения производных для того, чтобы можно было использовать высокоселективные и высокочувствительные детекторы (ЭЗД, ТИД). Однако это неизбежно сопровождается потерями анализируемого вещества в ходе этих операций, что приводит к увеличению ошибки анализа. Кроме этого свой вклад вносит также непостоянство состава анализируемой матрицы от пробы к пробе. В связи с этим химик-аналитик не всегда может удовлетворить желание гигиениста и токсиколога иметь МВИ с очень низкими пределами количественного определения вследствие технических возможностей используемых приборов и методических ограничений разрабатываемой МВИ. При разработке МВИ химик-аналитик свои усилия должен фокусировать не только на достижении низких пределов количественного определения анализируемых пестицидов, но не упускать из поля зрения более важные аспекты анализа остатков пестицидов: надежность идентификации и воспроизводимость результатов. Известно, что в настоящее время в Украине в некоторых сельскохозяйственных культурах и продуктах питания содержание пестицидов не допускается (так называемые zero tolerances) или находится на уровне предела обнаружения (limit of detection, LOD), т. е. любые детектируемые остатки пестицидов считаются недопустимыми. Для таких случаев первостепенное значение приобретает надежность идентификации пестицида, а не точное количественное определение его содержания, поскольку уже сам факт обнаружения пестицида является основанием для запрещения использования сельскохозяйственного сырья или продукта питания. В этих случаях применение полуколичественного варианта ТСХ является вполне оправданным при условии, что при этом достигается надежная идентификация определяемого пестицида.
Понимая, какое важное значение в анализе остатков пестицидов имеют вопросы, связанные с повышением надежности идентификации определяемых соединений, нами были предприняты систематические исследования по изучению межмолекулярных взаимодействий хлор- и азотсодержащих пестицидов в условиях газовой и жидкостной хроматографии. При этом было впервые установлено существование корреляционных зависимостей между параметрами удерживания членов гомологических рядов сорбатов, полученных при использовании хроматографических методов с различными механизмами сорбции. Эффективность использования таких зависимостей для повышения надежности идентификации пестицидов была продемонстрирована на примере гомологических рядов хлоралканкарбоновых и хлорфеноксиалканкарбоновых кислот и их эфиров, хлорфенолов, замещенных фенилмочевин, нитрофенолов и нитрофенольных соединений, замещенных бензойных кислот, симм-триазинов, эфиров тиокарбаминовой кислоты. [9]
Глава 3. МЕТОДИЧЕСКИЕ УКАЗАНИЯ ПО ОПРЕДЕЛЕНИЮ ХЛОРОРГАНИЧЕСКИХ ПЕСТИЦИДОВ В ВОДЕ, ПРОДУКТАХ ПИТАНИЯ, КОРМАХ И ТАБАЧНЫХ ИЗДЕЛИЯХ ХРОМАТОГРАФИЕЙ В ТОНКОМ СЛОЕ
Данная методика апробирована и рекомендована в качестве официальной группой экспертов при Госкомиссии по химическим средствам борьбы с вредителями, болезнями растений и сорняками при МСХ СССР.
Настоящие Методические указания распространяются на определение содержания ДДТ, ДДЭ, ДДД, гексохлорана, альдрина, кельтана, гептахлора, метоксихлора, дактала, тедиона и эфирсульфоната в воде, почве, вине, овощах, фруктах, грибах, зерне, комбикормах, корнеклубнеплодах и зеленых кормах, рыбе, мясе, мясопродуктах, внутренних органах, молоке и молочных продуктах, животном жире, сливочном и растительных маслах, жмыхах, шротах, лузге, меде, сахаре, яйцах и яйцепродуктах, а также в табачных изделиях.
Принцип метода. Метод основан на хроматографии хлорсодержащих пестицидов в тонком слое окиси алюминия, силикагеля или пластинок "Силуфол" в различных системах подвижных растворителей после экстракции их из исследуемых образцов и очистке экстрактов. Подвижным растворителем служит н-гексан или н-гексан в смеси с ацетоном. Места локализации препаратов обнаруживают после опрыскивания пластинок раствором аммиаката серебра с последующим ультрафиолетовым облучением или после облучения ультрафиолетовым светом пластинок "Силуфол", содержащих о-толидин.
Реактивы и растворы
Ацетон хч, ГОСТ 2603-71
Аммиак водный хч, ГОСТ 3760-64
Алюминия окись 2 ст. активности для хроматографии, ч, МРТУ 6-09-5296-68. Просеивают через сито 100 меш.
Алюминия окись, пропитанная серной кислотой. Две весовые части окиси алюминия (или окиси кремния) помещают в фарфоровую ступку, заливают одной объемной частью серной кислоты и тщательно перемешивают. Смесь готовят непосредственно перед подготовкой колонок для очистки экстрактов из проб шротов, жмыха, лузги
Бензол хч, ГОСТ 5955-68
Н-гексан ч, МРТУ 6-09-2937-66
Калий щавелевокислый чда, ГОСТ 5868-68
Кальций сернокислый чда, ГОСТ 3210-66. Просушивают 6 часов в сушильном шкафу при 160 град. C. Просеивают через сито 100 меш.
Кремния окись для люминофоров ч, МРТУ 6-09-4875-67
Натрий сернокислый безводный ч, ГОСТ 4166-66
Натрий углекислый кислый хч, ГОСТ 4201-66, 0,5 н. раствор
Натрий хлористый хч, ГОСТ 4233-66, насыщенный раствор
Петролейный эфир (темп. кип. 40 - 70 град.)
Перекись водорода хч (30% водный раствор), ГОСТ 10929-64
Проявляющие реактивы:
Проявляющий реактив N 1. 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, прибавляют 7 мл аммиака и доводят объем раствора до 100 мл ацетоном; в готовый раствор можно добавить 0,2 мл перекиси водорода. Раствор следует хранить в колбе с притертой пробкой в темном месте в течение 3-х дней. На пластинку 9 x 12 см расходуется 8 - 10 мл раствора. Проявляющий реактив N 2. 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, добавляют 10 мл 2-феноксиэтанола и доводят объем раствора до 200 мл ацетоном, затем добавляют 6 капель 30-процентной перекиси водорода.
Серебро азотнокислое чда, ГОСТ 1277-63
Серная кислота ч, ГОСТ 4204-66
Силикагель АСК (Воскресенского химкомбината Московской обл.)
Силикагель КСК, просеянный через сито 100 меш.
Стандартные образцы:
ДДТ, ДДД, ДДЭ, альдрин, изомеры ГХЦГ, гептахлор, метоксихлор, кельтан, эфирсульфонат, дактал, тедион хч.
Стандартные растворы: 10 мг соответствующего пестицида растворяют в мерной колбе на 100 мл в н-гексане и доводят до метки этим растворителем. Стандартные растворы необходимо хранить в стеклянной посуде с притертыми пробками в холодильнике.
Стеклянная вата, очищенная конц. серной кислотой, промытая дистиллированной водой и высушенная о-Толидин ч, МРТУ 6-09-6337-69, 1% раствор в ацетоне2-феноксиэтанол
Этиловый спирт, ректификат, ТУ 19-11-39-69
Хлороформ хч, ГОСТ 200-15-74
Четыреххлористый углерод хч, ГОСТ 20228-74
Этиловый эфир (для наркоза), Фармакопея СССР
Натрий сернокислый, 2% водный раствор
Натрий сернокислый, насыщенный раствор
2.4. Приборы и посуда
Баня водяная, ТУ 64-1-2850-76
Вакуумно-ротационный испаритель, ИР ТУ 25-11-310-69 или прибор для отгонки растворителей, МРТУ 25-11-67-67
Воронки химические, диам. 6 см, ГОСТ 86-13-64
Воронки делительные, емкостью 100, 250, 500 мл, ГОСТ 10054-75
Воронки Бюхнера, ГОСТ 9147-69
Гомогенизатор или измельчитель тканей
Камера для опрыскивания, ТУ 25-11-430-70
Камера для хроматографирования, размером 150 x 200, 105 x 165 мм, ГОСТ 10565-63
Колбы Бунзена, ТУ 25-11-135-69
Колбы мерные, емкостью 50, 100 мл, ГОСТ 1770-74
Колбы нш, емкостью 100, 250, 500 мл, ГОСТ 10394-63
Колбы круглодонные нш, емкостью 150, 250, 500 мл, ГОСТ 10394-63
Микропипетки, ГОСТ 1770-74 (для нанесения стандартных растворов)
Пипетки или шприцы для нанесения проб
Пипетки емкостью 1, 5, 10 мл, ГОСТ 1770-74
Прибор для встряхивания, МРТУ 2451-64
Пластинки стеклянные 9 x 12, 13 x 18 см
Пульверизаторы стеклянные для опрыскивания пластинок
Сито на 100 меш (диаметр отверстий 0,147 мм)
Стеклянные хроматографические колонки (диаметр - высота), 20 x 400, 15 x 150
Ртутно-кварцевая лампа
Цилиндры мерные емкостью 25, 50, 100, 250, 500 мл, ГОСТ 1770-74
Чашки выпарительные N 3, N 1, ГОСТ 9147-69
Приготовление пластинок для хроматографии
Тщательно промытую хромовой смесью, раствором соды, дистиллированной водой и высушенную пластинку протирают этиловым спиртом или эфиром и
покрывают сорбционной массой. Массу готовят следующим образом:
а) 50 г просеянной через сито 100 меш. окиси алюминия смешивают в фарфоровой ступке с 5 г сернокислого кальция, прибавляют 75 мл
дистиллированной воды и перемешивают в ступке или колбе до образования однородной массы. На пластинку 9 x 12 см наносят 10 г сорбционной массы (на пластинку 13 x 18 см - 20 г) и, покачивая, равномерно распределяют по всей пластинке. Пластинки сушат при комнатной температуре 18 - 20 часов, можно сушить их 20 минут при комнатной температуре, а затем 45 минут в сушильном шкафу при температуре 110 град. C.
б) 35 г силикагеля КСК, просеянного через сито 100 меш., смешивают с 2 г сернокислого кальция и 90 мл дистиллированной воды и перемешивают в ступке или колбе до однородной массы. Наносят на пластинки и сушат, как указано выше. Порция рассчитана на 10 пластинок.
Если пластинки с тонким слоем силикагеля темнеют после облучения УФ-светом, силикагель перед употреблением следует очистить от примесей. Для этого силикагель заливают на 18 - 20 часов разбавленной соляной кислотой (1:1), кислоту сливают, промывают силикагель водой и кипятят в круглодонной колбе 2 - 3 часа с разбавленной азотной кислотой (1:1), промывают проточной водопроводной, затем дистиллированной водой до нейтральной реакции промывных вод, сушат в сушильном шкафу 4 - 6 часов при температуре 130 град. Силикагель дробят и просеивают через сито 100 меш.
Пластинки для хроматографии "Силуфол" UV-254 производства ЧССР перед использованием импрегнируют о-толидином. Для этого каждую пластинку опускают на 0,5 см в 0,1% раствор о-толидина в ацетоне, налитый в камеру для хроматографирования. После того как фронт растворителя поднимется до верхнего края пластинки, ее вынимают и высушивают на воздухе, избегая прямого солнечного света. После этого пластинки готовы к употреблению. Пластинки, импрегнированные о-толидином, хранят в эксикаторе. Используют при анализе кормов.
Пластинки "Силуфол" UV-254 производства ЧССР предварительно промывают дистиллированной водой в хроматографической камере, высушивают на воздухе и непосредственно перед использованием активизируют в сушильном шкафу при температуре 65 град. в течение 4 минут. Подготовка хроматографических колонок для очистки экстрактов
Хроматографическая колонка для очистки от молочного жира. В нижнюю часть хроматографической колонки (размером 20 x 400 мм) помещают стекловату или 500 мг обезжиренной ваты. Затем засыпают в колонку силикагель АСК (75 мл для очистки экстрактов из проб свиного жира и 70 мл для всех остальных проб) и уплотняют силикагель постукиванием по колонке. Колонку промывают 50 мл н-гексана или петролейного эфира, и прошедший через нее растворитель отбрасывают. После этого колонка готова для хроматографической очистки экстрактов из проб рыбы, мяса и мясопродуктов, молока и молокопродуктов, меда, яиц и т.д.
Хроматографическая колонка для очистки экстрактов из проб шротов (необогащенных липидами) и лузги.
Хроматографическую колонку заполняют на высоту 1 см стеклянной ватой, затем в колонку вносят просеянную окись алюминия (I) слоем 2,5 см или окись кремния - 3,5 см. Далее засыпают, не утрамбовывая, комочки окиси алюминия (кремния), пропитанные серной кислотой, высота слоя (II) 2,5 см. Каждый слой последовательно промывают гексаном (всего 20 - 30 мл).
Для анализа жмыхов и шротов, обогащенных липидами, слоиокиси алюминия следует увеличить до 5 см (I) и 3 см (II) соответственно, в случае использования окиси кремния - 6 см (I) и 3 см (II).
Вода, вино. 200 мл пробы помещают в делительную воронку и экстрагируют пестициды, встряхивая в течение 3-х минут н-гексаном или петролейным эфиром тремя порциями по 30 мл или диэтиловым эфиром тремя порциями по 50 мл. В объединенные экстракты насыпают 10 г безводного сернокислого натрия или фильтруют через воронку, заполненную на 2/3 сернокислым натрием. Экстракты переносят в прибор для отгонки растворителей и отгоняют растворитель до объема 0,2 - 0,3 мл. В случае необходимости экстракт чистят серной кислотой.
Овощи, фрукты. 20 г измельченной пробы помещают в колбу с притертой пробкой и проводят экстрагирование пестицидов трижды в течение 15 минут на аппарате для встряхивания н-гексаном или петролейным эфиром порциями по 30 мл. Объединенные экстракты сушат безводным сернокислым натрием, переносят в прибор для отгонки растворителей, отгоняют растворитель до объема 0,2 - 0,3 мл и наносят на пластинку.
Зерно, грибы. Из измельченных проб отбирают 20 г зерна, 50 г сырых или 10 г сухих грибов и помещают в колбы с притертыми пробками. Экстракцию пестицидов проводят трижды на приборе для встряхивания н-гексаном или петролейным эфиром порциями по 30 мл. Объединенные экстракты переносят в делительную воронку, прибавляют 10 мл насыщенного раствора безводного сернокислого натрия в серной кислоте и осторожно встряхивают несколько раз. Отделяют органический слой и повторяют обработку до тех пор, пока кислота не станет бесцветной. Экстракт промывают дистиллированной водой, сушат безводным сернокислым натрием и отгоняют растворитель.
Яблоки, капуста, трава, сено. 20 г измельченных яблок, 20 г капусты, 40 г травы и 20 г сена заливают 100 мл ацетона в колбе с притертой пробкой. Встряхивают 2 - 3 минуты, прибавляют 20 мл дистиллированной воды и охлаждают на льду 30 минут. Экстракт сливают и фильтруют холодным, экстракцию повторяют. Из объединенных водно-ацетоновых экстрактов отгоняют ацетон, а из водного остатка экстрагируют препараты н-гексаном тремя порциями по 10 мл в течение 10 минут. Гексановые экстракты очищают серной кислотой, насыщенной безводным сернокислым натрием. Сушат безводным сернокислым натрием. Отгоняют растворитель до небольшого объема и наносят на пластинку. Если очистка неполная (после испарения растворителя на колбе остается белый налет), экстракт испаряют досуха, остаток смывают холодным ацетоном 3 раза порциями по 0,2 мл и сразу наносят на пластинку.
Комбикорма. Для исследования берут навеску 40 г, увлажняют ее в колбе 60 мл дистиллированной воды. Увлажненную навеску оставляют на ночь в колбе, закрытой пробкой. Экстракцию пестицидов проводят дважды 50 - 100 мл смеси гексан - ацетон 1:1 при встряхивании в течение 2 часов. Экстракты объединяют в делительной воронке на 500 мл, прибавляют дважды по 50 мл дистиллированной воды и, после разделения слоев, нижний водный слой сливают в другую делительную воронку и экстрагируют пестициды 40 мл гексана. Водный слой сливают. Гексановые экстракты объединяют, фильтруют через воронку с бумажным фильтром, заполненным на 2/3 безводным сернокислым натрием. Экстракты упаривают на ротационном испарителе до объема 20 - 30 мл или досуха, растворяя затем сухой остаток в 20 - 30 мл гексана или петролейного эфира. Экстракт переносят в делительную воронку и производят очистку серной кислотой, как описано выше.
Шрот, лузга, жмых. Навески: 15 г обогащенного липидами шрота или жмыха; 20 г не обогащенного липидами шрота или лузги делят на равные части и помещают в колбы емкостью 100 - 250 мл с притертыми пробками, заливают гексаном (три объема гексана на одну весовую часть шрота), встряхивают на приборе для встряхивания 30 минут. Экстракт фильтруют через воронку Бюхнера, не перенося осадок на воронку. В колбу повторно заливают указанное количество гексана, встряхивают 30 минут, фильтруют, количественно переносят осадок на воронку Бюхнера с помощью 30 мл гексана (3 раза по 10 мл). Полученный экстракт выпаривают до 30 мл на ротационном испарителе или в токе воздуха при температуре не выше 40 град., остаток делят на две равные части и помещают в морозильную камеру холодильника на 1 час (не менее). Каждую часть пропускают через отдельную колонку с окисью алюминия со скоростью 2 мл/минуту, промывают колбу и колонку 50 мл охлажденной смеси этилового эфира с гексаном (15:85). Эту операцию необходимо проводить без перерыва, не оставляя на следующий день. Очищенные экстракты объединяют и упаривают до объема 1 мл. Остаток из колбы переносят количественно микропипеткой с помощью резиновой груши в пробирку на 1 мл, колбу и микропипетку 2 - 3 раза промывают небольшим количеством гексана (всего 0,3 - 0,5 мл), сливая его в ту же пробирку. Затем осторожно выпаривают гексан из пробирки на водяной бане при температуре 50° почти досуха (конечный объем приблизительно 2 - 3 капли). Если общий объем экстракта и промывной жидкости превышает 1 мл, то сначала выпаривают экстракт, постепенно прибавляя к нему промывную жидкость. При наличии в упаренном экстракте белого мазеобразного осадка в пробирку добавляют 5 - 6 капель гексана и помещают ее на 15 - 20 минут в морозильную камеру холодильника, затем деканируют дважды таким же количеством гексана и снова упаривают до конечного объема 2 - 3 капли.
Параллельно с исследуемыми образцами готовят два модельных экстракта. Каждый экстракт получают из одного грамма шрота, не содержащего пестицидов (соотношение сухого вещества и пестицида то же, что и в исследуемых образцах). В один из экстрактов перед очисткой на колонках вносят микрошприцем (микропипеткой) определяемые пестициды в количестве 3 мкг, в другой - 0,75 мкг. Упаренные исследуемые и модельные экстракты с помощью микропипетки или микрошприца количественно наносят на пластинку, трижды смывая пробирку небольшим количеством гексана.
Рыба, мясо и мясопродукты. Мясо, мясопродукты пропускают через мясорубку. Рыбу очищают от чешуи, внутренних органов и тоже пропускают через мясорубку. 20 г пробы перемешивают с безводным сернокислым натрием и помещают в колбу с притертой пробкой. Пестициды экстрагируют дважды смесью гексан - ацетон или петролейный эфир - ацетон в соотношении 1:1 порциями по 50 мл в течение 1,5 часов при встряхивании.
Экстракт фильтруют через воронку с бумажным фильтром, заполненным на 2/3 безводным сернокислым натрием, затем растворитель отгоняют, сухой остаток растворяют в 20 мл н-гексана и вносят его в колонку с силикагелем АСК. После впитывания экстракта в сорбент пестицид элюируют 110 мл смеси бензола с гексаном в соотношении 3:8 порциями по 25 - 30 мл. Элюат собирают в круглодонную колбу со шлифом емкостью 250 - 300 мл. Через 10 минут после впитывания последней порции растворителя сорбент отжимают с помощью груши. Элюат отгоняют до объема 0,1 мл и наносят на хроматографическую пластинку.
В том случае, если пробы мяса или рыбы содержат большое количество жира, после испарения первого экстрагента (смеси ацетона с гексаном) и растворения сухого остатка в гексане следует провести очистку гексанового экстракта серной кислотой, а затем колоночную очистку, как описано выше.
Животный жир, яйца, яичный порошок. Жир измельчают на мясорубке, яичный порошок тщательно перемешивают, яйца - отделяют желток от белка, взвешивают желток и белок, а для анализа берут только желток. Конечный результат о содержании хлорорганических пестицидов в яйце приводят на все яйцо. Желток тщательно перемешивают. 25 г подготовленного образца заливают 50 мл ацетона, перемешивают и нагревают на горячей водяной бане до закипания растворителя. Колбу охлаждают, добавляют в нее 10 мл охлажденного 2% раствора сернокислого натрия, перемешивают и охлаждают 45 минут на ледяной бане. Затем сливают ацетоновый слой в круглодонную колбу через слой обезжиренной ваты. Экстракцию ацетоном с последующим вымораживанием жира повторяют еще два раза. Из объединенных экстрактов отгоняют ацетон на ротационном испарителе или в приборе для отгонки растворителей (температура бани не более 70 град. +/- 2 град.) и трижды экстрагируют петролейным эфиром порциями 20, 10 и 10 мл. Продолжительность первой экстракции 1 час, последующих - 15 минут. Петролейный эфир переносят в делительную воронку с 40 мл 2% водного раствора сернокислого натрия, перемешивают содержимое в течение 2-х минут, дают слоям разделиться и водную фазу отбрасывают. Для улучшения разделения слоев можно добавить несколько мл насыщенного раствора сернокислого натрия. Операцию промывки экстракта повторяют еще два раза, после чего петролейный эфир сливают в стакан с 20 г безводного сернокислого натрия, споласкивают делительную воронку дважды 5 мл петролейного эфира. Подсушенный экстракт количественно переносят в мерный цилиндр на 50 мл и доводят объем раствора петролейным эфиром до 30 мл. Далее наносят 30 мл экстракта в колонку с силикагелем АСК, как указано выше. Для проб свиного жира насыпают 75 мл силикагеля АСК, для всех остальных проб - 70 мл. Очистку экстрактов проводят, как описано для проб мяса. Элюат собирают в круглодонную колбу на 150 мл, растворитель упаривают до объема нескольких капель и наносят на хроматографическую пластинку.
Мед. 30 г меда смешивают с 3 г безводного сернокислого натрия и трижды экстрагируют пестициды гексаном порциями по 30 мл каждый раз по 15 минут, тщательно растирая мед стеклянной палочкой в узком химическом стакане. Экстракты объединяют и отгоняют гексан до объема 30 мл или до меньшего объема, далее доводя экстракт до 30 мл гексаном. 30 мл экстракта вносят в хроматографическую колонку с силикагелем АСК и проводят очистку экстракта и испарение растворителя, как описано выше.
Сахар. Из навески 50 г сахара, предварительно растворенного в воде, пестициды экстрагируют в делительной воронке на 250 мл н-гексаном. Экстракцию пестицидов проводят трижды по 50, 25 и 25 мл растворителя каждый раз встряхивая по 5 минут. Объединенные гексановые экстракты очищают от коэкстрактивных веществ (красящие, аминокислоты, липиды) сернокислотным способом.
Молоко и молочные продукты. Для подготовки проб можно использовать один из приведенных способов.
Первый способ. Сливки, сметана, молоко и др. цельномолочные продукты. Для анализа берут 20 г сливок и сметаны, предварительно разведенных равным объемом дистиллированной воды, 50 мл молока, кефира и т.п., прибавляют концентрированную серную кислоту (30 - 40 мл) до полного почернения пробы. Охлажденный до 10 - 15 град. раствор переносят в делительную воронку и экстрагируют препараты гексаном 2 раза порциями по 25 мл. Для полного извлечения воронку встряхивают 2 минуты, затем оставляют ее на 30 минут до полного разделения слоев. Если образуется эмульсия, прибавляют 1 - 2 мл этилового спирта. К объединенным экстрактам в делительной воронке прибавляют 10 мл концентрированной серной кислоты, насыщенной сернокислым натрием, и осторожно встряхивают несколько раз. Очистку продолжают до получения бесцветной серной кислоты.
Творог, сыр. 50 г творога или 10 г измельченного на терке сыра заливают 40 мл гексана или петролейного эфира, непрерывно встряхивают 2 - 3 минуты и оставляют на 30 минут. Экстракцию повторяют. Объединенные экстракты в делительной воронке очищают серной кислотой, как указано выше.
Второй способ. Молоко, кефир, простокваша, кумыс и другие цельномолочные продукты. 25 мл продукта помещают в делительную воронку на 300 мл, приливают по 5 мл щавелевокислого калия и насыщенного раствора хлористого натрия, перемешивают, приливают 100 мл ацетона, встряхивают 2 минуты. Приливают 100 мл хлороформа и встряхивают 2 мин. Воронку оставляют до полного разделения слоев. Верхнюю фазу отбрасывают, а нижнюю выливают в круглодонную колбу со шлифом и испаряют растворитель досуха. Остаток смывают 30 мл гексана.
Сгущенное молоко, 10 - 20% сливки. К 10 г продукта прибавляют 10 мл насыщенного раствора хлористого натрия и выливают в делительную воронку вместимостью 150 мл. К смеси приливают 40 мл ацетона, встряхивают 2 минуты, приливают 60 мл хлороформа, встряхивают 2 - 3 минуты и оставляют до разделения фаз. Далее поступают, как при определении пестицидов в молоке.
Сгущенные молочные продукты. 10 г продукта помещают в стаканчик, заливают 10 мл воды с температурой 45 - 50 град. C, перемешивают и переносят в делительную воронку на 150 мл, добавляют 5 мл щавелевокислого калия. Содержимое воронки перемешивают, приливают 80 мл ацетона и встряхивают 2 - 3 минуты. Добавляют 100 мл хлороформа и встряхивают 5 - 7 минут. После разделения фаз нижнюю фазу сливают в круглодонную колбу, растворители отгоняют, а сухой остаток растворяют в 30 мл петролейного эфира. Сухие молочные продукты. 3 г сухих молочных продуктов (сливок 2 г) высыпают в стаканчик, приливают 15 мл дистиллированной воды с температурой 40 - 45 град. C, размешивают и переносят в делительную воронку вместимостью 300 мл, приливают по 5 мл щавелевокислого калия и насыщенного раствора хлористого натрия. Содержимое воронки перемешивают, добавляют 80 мл ацетона и встряхивают 3 - 5 минут, приливают 100 мл хлороформа, встряхивают 5 минут и оставляют на 3 - 5 минут (до разделения фаз). Нижнюю фазу сливают в круглодонную колбу, растворитель отгоняют, а остаток смывают 30 мл гексана. Сметана, 30 - 40% сливки. 5 г продукта отвешивают в стаканчик, приливают 10 мл насыщенного раствора хлористого натрия и переносят в делительную воронку вместимостью 150 мл. Стаканчик обмывают 40 мл ацетона, смывы переносят в делительную воронку, которую встряхивают 2 - 3 минуты, добавляют 70 мл хлороформа и встряхивают 2 мин. Воронку оставляют на несколько минут до разделения фаз, нижнюю фазу сливают в колбу для отгонки растворителей, растворитель отгоняют, а остаток смывают 30 мл гексана.
Творог, сыр. 10 г творога или измельченного на терке сыра растирают с 10 мл насыщенного раствора хлористого натрия и переносят в делительную воронку на 250 - 300 мл. Прибавляют 80 мл ацетона, встряхивают 2 минуты, приливают 100 мл хлороформа и вновь встряхивают. Нижнюю фазу используют для анализа после отгонки растворителей, растворив остаток в 30
мл гексана. Далее проводят очистку экстрактов из проб молока и молочных продуктов от молочного жира, подготовленных по второму способу. Для этого 30 мл экстракта наносят в колонку с 70 мл силикагеля АСК. После впитывания экстракта в сорбент пестицид элюируют 110 мл смеси бензола с гексаном в соотношении 3:8 порциями по 25 - 30 мл. Элюат собирают в круглодонную колбу на 250 - 300 мл. Через 10 минут после впитывания последней порции растворителя сорбент отжимают с помощью резиновой груши. После очистки растворители отгоняют под вакуумом.
Сливочное масло. 20 г сливочного масла растапливают на водяной бане в круглодонной колбе, прибавляют 50 мл ацетона, тщательно перемешивают до растворения жира, прибавляют 10 мл ледянойдистиллированной воды и охлаждают на льду до затвердевания жира (примерно 30 минут). Сливают ацетоновый экстракт, и процедуру повторяют еще 2 раза. Из объединенных экстрактов в круглодонной колбе ацетон отгоняют на водяной бане. Пестициды экстрагируют из оставшегося водного экстракта гексаном тремя порциями по 10 мл в течение 5 минут. Объединенные гексановые экстракты в делительной воронке обрабатывают серной кислотой с сернокислым натрием. Очищенный экстракт сушат безводным сернокислым натрием и упаривают. Почва. К навескам воздушно-сухой почвы (10 г), помещенным в конические колбы емкостью 250 мл, приливают 10 мл 1-процентного водного раствора хлористого аммония и оставляют на сутки закрытыми. Затем приливают смесь 30 мл ацетона и 30 мл гексана и встряхивают колбы в течение часа на встряхивающем устройстве. Содержимое колб переносят в центрифужные пробирки. После центрифугирования жидкую часть сливают в конические колбы, почву с помощью 10 мл 1-процентного раствора хлористого аммония и 30 мл ацетона переносят в исходные конические колбы, добавляют 30 мл гексана и проводят экстракцию еще в течение 30 минут. Затем экстракты объединяют. К объединенным экстрактам в делительной воронке приливают180 мл дистиллированной воды, осторожно встряхивают в течение 5 - 7 минут, дают жидкостям расслоиться и нижний водный слой сливают в коническую колбу. Гексановый слой пропускают через безводный сульфат натрия (столовая ложка или 30 - 40 г сульфата натрия). Из водно-ацетонового слоя экстракцию пестицидов проводят еще дважды 15 и 10 мл гексана, который затем сушат через тот же сульфат натрия. Гексановые экстракты объединяют. Концентрирование экстрактов проводят либо на ротационно-вакуумном испарителе, либо при температуре бани не более 40 град. C и времени отгонки 9 - 11 минут, либо из колбочек с г-образным отводом при температуре водяной бани 72 - 75 град. C.
Очистку сконцентрированных гексановых экстрактов из проб почв проводят серной кислотой, как описано выше для других проб, и испаряют растворитель. Табак и табачные изделия. 5 г табака помещают в стеклянный стакан на 500 мл, заливают 50 мл концентрированной серной кислоты и стеклянной палочкой тщательно размешивают до полного равномерного обугливания пробы. Спустя 10 - 15 минут в колбу добавляют 25 мл гексана, тщательно размешивают содержимое и прибавляют 20 мл четыреххлористого углерода. Экстракцию пестицидов из пробы проводят в течение 15 минут трижды, после чего экстракт последовательно переносят в делительную воронку для однократной или двукратной дополнительной очистки серной кислотой.
Хроматографирование.
На хроматографическую пластинку на расстоянии 1,5 см от ее края шприцем или пипеткой наносят исследуемую пробу в одну точку так, чтобы диаметр пятна не превышал 1 см. Остаток экстракта в колбочке смывают тремя порциями (по 0,2 мл) диэтилового эфира, которые наносят в центр первого пятна. Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, с содержащие 10, 5, 1 мкг исследуемых препаратов (или другие количества, близкие к определяемым концентрациям).
Пластинки с нанесенными растворами помещают в камеру для хроматографирования, на дно которой за 30 минут до начала хроматографирования наливают подвижный растворитель. При использовании пластинок с тонким слоем окиси алюминия или силикагеля в качестве подвижного растворителя применяют н-гексан или смесь гексана с ацетоном в соотношении 6:1, для препаратов, у которых величина R в гексане ниже 0,3. При использовании f пластинок "Силуфол" подвижный растворитель - 1% раствор ацетона в гексане, а пластинок "Силуфол", импрегнированных о-толидином, - гексан с диэтиловым эфиром в соотношении 49:1. Край пластинки с нанесенными растворами может быть погружен в подвижный растворитель не более чем на 0,5 см.
После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Далее пластинку орошают проявляющим реактивом и подвергают действию УФ света в течение 10 - 15 минут (лампа ПРК-4). Пластинки следует располагать на расстоянии 20 см от источника света.
При наличии хлорорганических пестицидов на пластинке появляются пятна серо-черного цвета. При использовании для анализа пластинок "Силуфол", импрегнированных о-толидином, их непосредственно после хроматографирования подвергают УФ-облучению в течение нескольких минут. При наличии хлорорганических пестицидов в этом случае появляются пятна сине-голубого цвета. Количественное определение осуществляют сравнением площадей пятен пробы и стандартных растворов. Между количеством препарата в пробе, не превышающим 20 мкг, и площадью его пятна на пластинке существует прямая пропорциональная зависимость. При большем содержании препарата следует использовать пропорциональную часть исследуемого экстракта.
Глава 4. СОВРЕМЕННОН АППАРАТУРНОЕ ОФОРМЛЕНИЕ
СИСТЕМА ДЛЯ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ С ДЕНСИТОМЕТРОМ "ДенСкан"
Назначение и область применения
Системы для тонкослойной хроматографии и электрофореза с денситометром "ДенСкан" предназначены для качественного и количественного анализа состава проб веществ и материалов в видимой области спектра и ультрафиолетовом свете при длинах волн 254 и 365 нм.
Область применения - исследования в химии, биохимии, биологии, медицине, фармакологии, аналитическом контроле чистых веществ, объектов окружающей среды и др.
Технические данные
· Денситометр обеспечивает расчет параметров и количественную оценку хроматограмм в видимой и ультрафиолетовой области спектра ( lmax= 254 нм, lmax =365 нм)
· Размер обрабатываемых пластин, см .................................. не более 15 х 15
· Время ввода изображения, с .................................. .......... не более 5
· Время обмера хроматограммы, мин ................................... ………5
· Отношение сигнал/шум : видимая область............ не менее 5/1
· УФ, 254 нм ...................................................................... не менее 5/1
· УФ, 365 нм .............................................................. не менее 5/1
· Относительное СКО по площади пятен, %
· видимая область................................................................ не более 5
· УФ, 254 нм ....................................................................... не более 5
· УФ, 365 нм ....................................................................... не более 5
· Размах значений Rf : видимая область .......... не более 0,02
· УФ, 254 нм .............................................................. не более 0,02
· УФ, 365 нм ............................................................... не более 0.02
· Масса осветительной камеры, кг .............................. не более 12 кг
· Габаритные размеры осветительной камеры, мм .... не более длина ............................................................................... 420
ширина ............................................................................. 420
высота .............................................................................. 700
· Напряжение питания, В ............................................ 220 ± 22/33
· Частота переменного тока, Гц ............................................ 50 ± 1
· Средняя наработка на отказ денситометра, ч .... не менее 5000
Состав денситометра
Денситометр "ДенСкан" состоит из камеры осветительной, черно-белой либо цветной видеокамеры или сканнера, блока ввода изображения, системы обработки данных.
Камера осветительная выполнена в виде блочной конструкции, включающей следующие основные узлы:
- источники света:
лампы дневного света
лампы УФ диапазона, длина волны 254 нм
лампы УФ диапазона, длина волны 365 нм
- набор корректирующих светофильтров
- детектор - черно-белая малогабаритная видеокамера OS-45D или аналогичная с чувствительностью не хуже 0,02 люкс, с ручной фокусировкой и ручной регулировкой диафрагмы либо цветной сканнер с разрешением от 200 d.p.i. и выше с интерфейсом, соответствующим TWAIN стандарту
- установочный столик для пластин
- канал связи с блоком ввода изображения
Система обработки данных с использованием персонального компьютера и программного обеспечения "Dens". Минимальные требования к компьютеру:
- Операционная система - Microsoft Windows 95, Windows 98, Windows NT (версия 4.0 или выше)
- Процессор - Pentium 100 MHz
- Оперативная память (RAM) - 8 Мбайт, рекомендуется 16 Мбайт
- Цветной монитор - с диагональю не менее 14 дюймов
- Место на жестком диске - 10 Мбайт
- Манипулятор - "мышь"
Блок ввода изображения видеобластер AverMedia (и программное обеспечение к нему) используется для получения изображения хроматограммы на мониторе компьютера. Возможно использование аналогичных систем.
Пластины и листы для тонкослойной хроматографии (TLC)
| Код |
Описание |
| S0002020 |
стеклянные пластины для TLC: SIL G-25, 20×20 см., 25 шт. |
| S0UV2020 |
стеклянные пластины для TLC: SIL G-25 UV 254, 20×20 см., 25 шт. |
| SP002020 |
листы из полиэстера: SIL G, 20×20 см., 25 шт. |
| SPUV2020 |
листы из полиэстера: SIL G/UV 254, 20×20 см., 25 шт. |
| SA002020 |
алюминиевые листы: SIL G, 20×20 см., 25 шт. |
| SAUV2020 |
алюминиевые листы: SIL G/UV 254, 20×20 см., 25 шт. |
Шприц для хроматографии МШ-50 (М-50) (шток из нержавеющей стали, с противооткатной муфтой) Шприц для хроматографии М-1Н (МШ-1), М-5Н (с направляющей)
Шприц для хроматографии МШ-10 (М-10Н), МШ-50 (М-50Н) (шток из нержавеющей стали, с направляющей)
Шприц для хроматографии МШ-10М (М-10) (шток из нержавеющей стали, с противооткатной муфтой)[10]
Литература
1. Кирхнер Ю. Тонкослойная хроматография. М.: Мир, 1981.
2. Хроматография в тонких слоях / Под ред. Э. Шталя. М.: Мир, 1965.
3. Евгеньев М.И., Евгеньева И.И., Москва Н.А., Левинсон Ф.С. 5-Хлор-4,6-динитробензофуразан как реагент в тонкослойной хроматографии ароматических аминов // Завод. лаб. 1992. Т. 58, № 4. С. 11-13.
4. Назаркина С.Г. Определение полиароматических углеводородов в объектах окружающей среды методами жидкостной и тонкослойной хроматографии.
5. Соголовский Б.М. Денситометр «Сорбфил» для количественной ТСХ
6. Руководство по химическому анализу поверхностных вод суши (под редакцией А.Д. Семенова) // Л.: Гидрометеоиздат. - 1977. - 540 с.
7. Унифицированные методы анализа вод. Под редакцией Ю.Ю. Лурье // М.:Химия. - 1973. - 376 с.
8. Лурье Ю.Ю. Аналитическая химия промышленных и сточных вод. // М.: Химия. - 1984. - 447 с.
9. В.Д. Чмиль Состояние и перспективы использования современных инструментальных методов анализа пестицидов в Украине
10. http://www.izme.ru/
|