Курсовая работа
На тему
«Хроматографические методы анализа и их использование в анализе объектов окружающей природной среды»
Содержание
Введение
Глава 1. Хроматография в современной химии
1.1. Основные виды хроматографии
1.2. Методы проявления хроматограмм
1.3. Работа хроматографа
Глава 2. Применение хроматографических методов в экологическом мониторинге
2.1. Аппаратура для хроматографии
Глава 3. Примеры применения хроматографии в анализе объектов окружающей среды
Глава 4. Современное аппаратурное оформление
Литература
Введение
Исключительно мощное средство контроля загрязнения различных объектов окружающей среды - хроматографические методы, позволяющие анализировать сложные смеси компонентов. Наибольшее значение приобрели тонкослойная, газожидкостная и высокоэффективная жидкостная и ионная хроматография. Будучи несложной по технике выполнения, тонкослойная хроматография хороша при определении пестицидов и других органических соединений-загрязнителей. Газожидкостная хроматография эффективна при анализе многокомпонентных смесей летучих органических веществ. Применение различных детекторов, например малоизбирательного детектора по теплопроводности - катарометра и избирательных - пламенно-ионизационного, электронного захвата, атомно-эмиссионного, позволяет достигать высокой чувствительности при определении высокотоксичных соединений. Высокоэффективную жидкостную хроматографию применяют при анализе смесей многих загрязняющих веществ, прежде всего нелетучих. Используя высокочувствительные детекторы: спектрофотометрические, флуориметрические, электрохимические, можно определять очень малые количества веществ. При анализе смесей сложного состава особенно эффективно сочетание хроматографии с инфракрасной спектрометрией и особенно с масс-спектрометрией. В последнем случае роль детектора играет подключенный к хроматографу масс-спектрометр. Обычно приборы такого типа оснащены мощным компьютером. Так определяют пестициды, полихлорированные бифенилы, диоксины, нитрозоамины и другие токсичные вещества. Ионная хроматография удобна при анализе катионного и анионного составов вод.
Глава 1. Хроматография в современной химии
Одна из важных задач современной химии – надежный и точный анализ органических веществ, часто близких по строению и свойствам. Без этого невозможно проведение химических, биохимических и медицинских исследований, на этом в значительной степени базируются экологические методы анализа окружающей среды, криминалистическая экспертиза, а также химическая, нефтяная, газовая, пищевая, медицинская отрасли промышленности и многие другие отрасли народного хозяйства.
Один из наиболее чувствительных методов – хроматографический анализ, впервые предложенный российским ученым М.С.Цветом в начале XX в. и к концу века превратившийся в мощнейший инструмент, без которого уже не могут обходиться как синтетики, так и химики, работающие в других областях.
Разделение Цвет проводил в колонке, показанной на рис. 1. Смесь веществ А, Б и В – природных пигментов, первоначально находящихся в зоне е, – разделяется при приливании соответствующего растворителя Д (элюент) на отдельные зоны.

Рис. 1. Хроматографическое разделение пигментов хлорофилла М.C.Цветом: а – адсорбент; б – колонка; в – приемник; г – делительная воронка; д – вата.
Смесь веществ А, Б и В, сначала находящихся в зоне е, разделяется при элюировании растворителем Д (элюент) на отдельные зоны, движущиеся с разными скоростями к выходу из колонки.
Хроматография основана на распределении одного из нескольких веществ между двумя, как говорят, фазами (например, между твердым телом и газом, между двумя жидкостями и др.), причем одна из фаз постоянно перемещается, т. е. является подвижной.
Это значит, что такая фаза, например газ или жидкость, все время продвигается, нарушая равновесие. При этом чем лучше то или иное вещество сорбируется (поглощается) или растворяется в неподвижной фазе, тем скорость его движения меньше, и, наоборот, чем меньше сорбируется соединение, т. е. обладает меньшим сродством к неподвижной фазе, тем скорость перемещения больше. В итоге, как показано на рис. 2, если вначале мы имеем смесь соединений, то постепенно все они, подталкиваемые подвижной фазой, движутся к «финишу» с различными скоростями и в конце концов разделяются.

Рис. 2. Основной принцип хроматографического разделения: НФ – слой неподвижной фазы, покрывающей внутреннюю поверхность капиллярной трубки Т, через которую течет подвижная фаза (ПФ). Компонент А1
разделяемой смеси обладает большим сродством к подвижной фазе, а компонент А2
– к неподвижной фазе. А '1
и А '2
– положения зон тех же компонентов через промежуток времени, за которое происходило хроматографическое разделение в направлении, указанном стрелкой
Практически образец смеси веществ вводят, например, шприцем в слой неподвижной фазы, а затем различные соединения, входящие в состав смеси, вместе с подвижной фазой (элюент) двигаются вдоль слоя, подгоняемые этой фазой. Скорость перемещения зависит от величины взаимодействия (сродство) компонентов в неподвижной и подвижной фазах, и в результате достигается разделение компонентов.
После разделения необходимо идентифицировать все компоненты и оценить их количественно. Такова общая схема хроматографии.
Следует отметить, что этот современный метод позволяет в течение нескольких минут определить содержание десятков и сотен различных соединений в смеси, причем даже в ничтожных, «следовых» количествах ~10–8%. [1-3]
Хроматографический способ анализа.
Хроматографические системы можно разделить по следующим принципам:
– агрегатное состояние подвижной и неподвижной фаз;
– геометрические характеристики системы;
– механизм взаимодействия между разделяемым веществом и фазами.
В качестве подвижной фазы используется газ или жидкость. В качестве неподвижной, или стационарной, фазы применяются твердые вещества или жидкости.
По расположению фаз хроматографические системы подразделяют на две группы: плоскостные и колоночные.
Последние, в свою очередь, разделяются на:
– насадочные, заполненные зернистым твердым материалом (мелкие шарики), либо являющимся разделительной средой, либо служащим носителем неподвижной жидкой фазы;
– капиллярные, внутренние стенки которых покрыты пленкой неподвижной жидкости или слоем твердого адсорбента (поглотитель).
Взаимодействие между разделяемым веществом и фазами хроматографической системы может осуществляться или на поверхности фазы, или в объеме. В первом случае хроматография называется адсорбционной, во втором – распределительной.
Механизмы разделения молекул в хроматографических системах чаще всего сводятся к следующим:
– неподвижная фаза физически поглощает (сорбирует) разделяемые вещества;
– неподвижная фаза химически взаимодействует с разделяемыми веществами;
– неподвижная фаза растворяет разделяемые вещества из раствора в несмешивающемся растворителе;
– неподвижная фаза имеет пористую структуру, затрудняющую диффузию молекул разделяемых веществ в этой фазе.
Хроматография, начавшись с самодельных устройств типа полоски бумаги, опущенной в растворитель, в настоящее время представлена сложнейшими инструментальными системами, основанными на современных точнейших, или прецизионных, принципах и оснащенными компьютерным обеспечением. Схема процесса хроматографирования, в сущности, очень проста и показана на рис. 3. Далее примерно в такой последовательности будет рассмотрен принцип работы хроматографа.
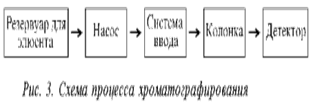
1.1 Основные виды хроматографии
К основным видам хроматографии относят адсорбционную, ионообменную, жидкостную, бумажную, тонкослойную, гель-фильтрационную и афинную хроматографию.
Адсорбционная хроматография
. В этом случае разделение веществ осуществляется за счет выборочной (селективной) адсорбции веществ на неподвижной фазе. Такая селективная адсорбция обусловлена сродством того или иного соединения к твердому адсорбенту (неподвижной фазе), а оно, в свою очередь, определяется полярными взаимодействиями их молекул. Поэтому часто хроматографию такого типа используют при анализе соединений, свойства которых определяются числом и типом полярных групп. К адсорбционной хроматографии причисляют ионообменную, жидкостную, бумажную, тонкослойную и газо-адсорбционную хроматографию.

Рис. 4. Изображение структуры частицы ионообменной смолы:
– заряженные функциональные группы, ковалентно связанные с нитями решетки;
– свободно перемещающиеся противоположно заряженные протовоионы, электростатически связанные с частицей смолы, способные претерпевать обмен с другими ионами.
Ионообменная хроматография.
В качестве неподвижной фазы используют ионообменные смолы (рис. 4) как в колонках, так и в виде тонкого слоя на пластинке или бумаге. Разделение обычно проводят в водных средах, поэтому этот метод используется главным образом в неорганической химии, хотя применяются и смешанные растворители. Движущей силой разделения в этом случае является различное сродство разделяемых ионов раствора к ионообменным центрам противоположной полярности в неподвижной фазе.
Жидкостная хроматография
. В этом случае неподвижной фазой служит жидкость. Наиболее распространенным случаем является адсорбционный вариант жидкостной колоночной хроматографии. Пример разделения природных пигментов представлен на рис. 5.

Рис. 5. Хроматографическое разделение природных пигментов (флавонов и изофлавонов)
Бумажная хроматография
. В качестве неподвижной фазы используют полосы или листы бумаги (рис. 6). Разделение происходит по адсорбционному механизму, причем иногда его проводят в двух перпендикулярных направлениях.

Рис. 6. Схема разделения методом бумажной хроматографии: А, Б и В – положения компонентов смеси по окончании хроматографического разделения. Подвижная фаза впитывается в бумагу (неподвижная фаза) под действием капиллярных сил и переносит индивидуальные компоненты смеси с различными скоростями, зависящими от отношений растворимости этих компонентов в обеих фазах. Отношение а/б = Rf
(фактор запаздывания) характеризует данное разделяемое вещество
Тонкослойная хроматография – это любая система, в которой неподвижной фазой является тонкий слой, в частности слой оксида алюминия (толщина 2 мм) в виде пасты, нанесенной на стеклянную пластинку. Пример такой системы и результаты разделения показаны на рис. 7.

Рис. 7. Камера для тонкослойной хроматографии:а – общий вид; б – схематический разрез; 1 – подложка со слоем сорбента; 2 – край камеры; 3 – емкость для растворителя
Гель-фильтрационная
, или молекулярно-ситовая, хроматография. Принцип разделения в таких системах несколько иной, чем в предыдущих случаях. Неподвижной фазой являются материалы, обычно гели, со строго контролируемой пористостью, в результате чего одни компоненты смеси в соответствии с размером и формой молекул могут проникать между частицами геля, а другие не могут. Наиболее часто этот вид хроматографии используется для разделения высокомолекулярных соединений. Один из вариантов применения этого метода – определение молекулярных масс разделяемых веществ, часто необходимых для химических исследований (рис. 8).

Рис. 8. Схема разделения методом гель-хроматографии: а – начало разделения б – разделение в – конец разделения; большие кружки – частицы геля, большие точки – молекулы соединений с большой молекулярной массой, маленькие точки – молекулы соединений с меньшей молекулярной массой
Афинная хроматография
. Этот вид хроматографии основан на взаимодействии между веществом, с одной стороны, способным реагировать с выделяемым соединением, а с другой – связанным с твердым носителем неподвижной фазы. Такое вещество обладает сродством к выделяемому соединению и называется афинным лигандом.
Наиболее часто этот метод находит применение в биохимическом анализе. Например, при пропускании через целлюлозу, активированную бромцианом, биологических объектов-антигенов, содержащих белки, происходит их специфическое удерживание, как показано на схеме 1.

По другому способу для присоединения белков к гидроксильной группе целлюлозы последнюю сначала обрабатывают 2-амино-4,6-дихлор-сим-триазином, а затем продукт их взаимодействия вступает в реакцию с аминогруппой белка по схеме 2:

Конечно, число способов хроматографирования не ограничивается перечисленными выше. Часто хроматографию сочетают с другими физико-химическими методами, например с масс-спектрометрией, но в данной статье стоит задача познакомить читателя лишь с общими принципами хроматографии. Поэтому далее рассмотрим обработку результатов хроматографирования.
1.2 Методы проявления хроматограмм
Проявлением называется процесс переноса разделяемых веществ подвижной фазой. Проявление можно осуществить тремя основными способами: фронтальным анализом, вытеснением и элюированием. Наиболее широко используется элюирование.
Фронтальный анализ. Это случай наиболее простой, т. к. здесь проба и служит подвижной фазой. Ее непрерывно добавляют в систему, поэтому нужны большие объемы пробы. Результаты показаны на рис. 9.

Образование нескольких зон обусловлено различным сродством разных компонентов к неподвижной фазе. Передний край называют фронтом, отсюда и название. В первой зоне находится только наименее удерживаемое вещество А, которое движется быстрее всего. Вторая зона содержит вещество А и Б. Третья зона – смесь веществ А, Б и В. Во фронтальном анализе только компонент А получают в жидком виде.
Вытеснительный анализ
. В этом случае подвижная фаза обладает большим сродством к неподвижной фазе, чем разделяемое вещество. В неподвижную фазу вводят небольшую пробу. Но из-за большого сродства подвижная фаза вытесняет и проталкивает все компоненты. Она вытесняет наиболее сильно сорбирующийся компонент В, который, в свою очередь, вытесняет вещество Б, а тот вытесняет наименее сорбирующийся компонент А. В отличие от фронтального анализа с помощью этого способа можно получить все основные компоненты в индивидуальном (жидком) виде.
Элюентный анализ
. Подвижную фазу для перемещения растворенного вещества пропускают через хроматографическую систему. Разделение происходит за счет различного сродства компонентов смеси к неподвижной фазе и, следовательно, за счет разных скоростей их перемещения. Пробу малого объема вводят в хроматографическую систему. В итоге зоны с компонентами будут постепенно образовывать отдельные участки, разделенные чистым элюентом. Благодаря высокой эффективности разделения метод получил наиболее широкое распространение и в значительной степени вытеснил другие варианты разделения. Поэтому далее рассмотрим теорию и аппаратное оформление этого метода.
Хроматографические процессы часто удобно рассматривать как серии экстракционных процессов, при этом могут быть разделены вещества с очень близкими свойствами, т. к. в ходе хроматографических процессов быстро и одновременно происходят сотни и даже тысячи циклов экстракции.
Для оценки эффективности хроматографических процессов, исходя из теоретического представления о дистилляции (по аналогии с разделением нефти на ректификационных колоннах, где теоретическая тарелка соответствует части ректификационной колонны, в которой пар и жидкость находятся в равновесии), вводят понятие «высота, эквивалентная теоретической тарелке» (ВЭТТ). Хроматографическая колонка, таким образом, рассматривается как набор гипотетических слоев (тарелок). Под ВЭТТ обычно подразумевают такую толщину слоя, которая необходима для того, чтобы смесь, поступившая из предыдущего слоя, пришла в равновесие со средней концентрацией вещества в подвижной фазе этого слоя. Ее можно описать следующей формулой:
ВЭТТ = L/N,
где L – длина колонки, N – число теоретических тарелок.
ВЭТТ является суммарной характеристикой разделения веществ. Однако разделить компоненты смеси важно, но недостаточно. Необходимо идентифицировать каждый компонент и определить его количество в пробе. Обычно это осуществляют с помощью обработки хроматограмм – зависимости интенсивности сигнала, пропорционального концентрации вещества, от времени разделения. Некоторые примеры хроматограмм показаны на рис. 9.

Время от момента ввода пробы в колонку до момента регистрации максимума пика называется временем удерживания (tR). В оптимальных условиях оно не зависит от количества введенной пробы и с учетом геометрических параметров колонки определяется строением того или иного соединения, т. е. является качественной характеристикой компонентов. Количественное содержание компонента характеризуется величиной пика, точнее его площадью. Подсчет площади пика обычно осуществляют автоматически с помощью прибора интегратора, который фиксирует и время удерживания, и площадь пика. Современная аппаратура позволяет сразу получать компьютерную распечатку с указанием содержания всех компонентов разделяемой смеси.

1.3 Работа хроматографа
Схема установки наиболее простого газового хроматографа приведена на рис. 12. Она состоит из газового баллона, содержащего подвижную инертную фазу (газ-носитель), чаще всего гелий, азот, аргон и др. С помощью редуктора, уменьшающего давление газа до необходимого, газ-носитель поступает в колонку, представляющую собой трубку, заполненную сорбентом или другим хроматографическим материалом, играющим роль неподвижной фазы.

Рис. 10. Схема работы газового хроматографа: 1 – баллон высокого давления с газом-носителем; 2 – стабилизатор потока; 3 и 3 ' – манометры; 4 – хроматографическая колонка; 5 – устройство для ввода пробы; 6 – термостат; 7 – детектор; 8 – самописец; 9 – расходомер
Хроматографическая колонка – это «сердце» хроматографа, поскольку именно в ней происходит разделение смесей. Колонки чаще всего изготавливают из стекла; бывают стальные, тефлоновые, а также капиллярные колонки. Вблизи от ввода газа в колонку устанавливают устройство для ввода пробы. Чаще всего вводят пробу с помощью шприца, протыкая резиновую мембрану. Анализируемая смесь разделяется в колонке и поступает в детектор – прибор, преобразующий результаты разделения в форму, удобную для регистрации.
Одним из наиболее используемых детекторов является катарометр, принцип действия которого основан на измерении теплоемкости разных тел.
На рис. 11 показана схема катарометра. В цилиндрическую полость помещена металлическая спираль (нить сопротивления), нагревающаяся в результате прохождения через нее постоянного электрического тока. При протекании через нее газа-носителя c постоянной скоростью температура спирали остается постоянной. Однако если состав газа меняется при появлении элюируемого вещества, то температура спирали меняется, что и регистрируется прибором.

Рис. 11. Схема катарометра: 1 – ввод газа из хроматографической колонки; 2 – вывод продуктов в атмосферу; 3 – нить сопротивления; 4 – изолятор; 5 – металлический блок катарометра
Другой распространенный детектор – пламенно-ионизационный, схема которого представлена на рис. 12. Он гораздо более чувствителен, чем катарометр, но требует подачи не только газа-носителя, но и водорода. Выходящий из колонки газ-носитель, содержащий элюент, смешивается с водородом и проходит в форсунку горелки детектора. Пламя ионизирует молекулы элюента, в результате чего электрическое сопротивление между электродами уменьшается, а ток увеличивается.

Рис. 12. Схема пламенно-ионизационного детектора: 1 – ввод водорода; 2 – ввод газа из хроматографической колонки; 3 – ввод воздуха; 4 – катод; 5 – горелка; 6 – собирающий электрод; 7 – вывод продуктов горения в атмосферу.
В жидкостной хроматографии применяются спектрофотометрические детекторы (в видимой, УФ - и ИК-областях), а также рефрактометрические детекторы, основанные на измерении показателей преломления растворов.
Глава 2. Применение хроматографических методов в экологическом мониторинге
Хроматографические методы
часто оказываются незаменимыми для идентификации и количественного определения органических веществ со сходной структурой. При этом наиболее широко используемыми для рутинных анализов загрязнителей окружающей среды являются газовая и высокоэффективная жидкостная хроматография.
Газохроматографический анализ органических загрязнителей в питьевой и сточных водах сначала основывался на использовании насадочных колонок, позднее распространение получили и кварцевые капиллярные колонки. Внутренний диаметр капиллярных колонок составляет обычно 0,20-0,75 мм, длина - 30-105 м. Оптимальные результаты при анализе загрязнителей в воде достигаются чаще всего при использовании капиллярных колонок с различной толщиной пленки из метилфенилсиликонов с содержанием фенильных групп 5 и 50%. Уязвимым местом хроматографических методик с использованием капиллярных колонок часто становится система ввода пробы. Системы ввода пробы можно подразделить на две группы: универсальные и селективные. К универсальным относятся системы ввода с делением и без деления потока, “холодный” ввод в колонку и испарение при программировании температуры. При селективном вводе используют продувку с промежуточным улавливанием в ловушке, парофазный анализ и т.д. При использовании универсальных систем ввода в колонку поступает вся проба полностью, при селективной инжекции вводится только определенная фракция. Результаты, получаемые при селективном вводе, являются существенно более точными, поскольку попавшая в колонку фракция содержит только летучие вещества, и техника при этом может быть полностью автоматизирована.
Газохроматографические детекторы, используемые в мониторинге загрязнителей, часто подразделяют на универсальные, откликающиеся на каждый компонент в подвижной фазе, и селективные, реагирующие на присутствие в подвижной фазе определенной группы веществ со сходными химическими характеристиками. К универсальным относятся пламенно-ионизационный, атомно-эмиссионный, масс-спектрометрический детекторы и инфракрасная спектрометрия. Селективными детекторами, используемыми в анализе воды, являются электронно-захватный (селективен к веществам, содержащим атомы галогенов), термоионный (селективен к азот- и фосфорсодержащим соединениям), фотоионизационный (селективен к ароматическим углеводородам), детектор по электролитической проводимости (селективен к соединениям, содержащим атомы галогенов, серы и азота). Минимально детектируемые количества веществ - от нанограммов до пикограммов в секунду.
Высокоэффективная жидкостная хроматография
(ВЭЖХ) является идеальным методом для определения большого числа термически неустойчивых соединений, которые не могут быть проанализированы с помощью газовой хроматографии. Объектами анализа методом жидкостной хроматографии в настоящее время часто становятся современные агрохимикаты, в число которых входят метилкарбонаты и фосфорорганические инсектициды, другие нелетучие вещества. Высокоэффективная жидкостная хроматография получает все большее распространение среди других методов, применяемых в мониторинге окружающей среды, еще и потому, что имеет блестящие перспективы в плане автоматизации пробоподготовки.
Колонки для ВЭЖХ, которые чаще всего используют в анализах загрязнителей окружающей среды, имеют длину 25 см и внутренний диаметр 4,6 мм, заполняются они сферическими частицами силикагеля размером 5-10 мкм с привитыми октадецильными группами. В последние годы появились колонки с меньшим внутренним диаметром, заполненными частицами меньшего размера. Использование таких колонок приводит к уменьшению расхода растворителей и продолжительности анализа, увеличению чувствительности и эффективности разделения, а также облегчает проблему подключения колонок к спектральным детекторам. Колонки с внутренним диаметром 3,1 мм снабжают предохранительным картриджем (форколонкой) для увеличения срока службы и улучшения воспроизводимости анализов.
В качестве детекторов в современных приборах для ВЭЖХ используются обычно УФ-детектор на диодной матрице, флуоресцентный и электрохимический.
Электроаналитические методы, которые обычно применяют в анализе воды для определения неорганических компонентов, часто уступают по чувствительности методам газовой и жидкостной хроматографии, атомно-адсорбционной спектрометрии. Однако здесь используется более дешевая аппаратура, иногда даже в полевых условиях. Основными электроаналитическими методами, применяемыми в анализе воды, являются вольтамперометрия,
потенциометрия и кондуктометрия.
Наиболее эффективными вольтамперометрическими методами являются дифференциальная импульсная полярография (ДИП) и инверсионный электрохимический анализ (ИЭА). Сочетание этих двух методов позволяет проводить определение с очень высокой чувствительностью - приблизительно 10-9
моль/л, аппаратурное оформление при этом несложно, что дает возможность делать анализы в полевых условиях. На принципе использования метода ИЭА или сочетания ИЭА с ДИП работают полностью автоматизированные станции мониторинга. Методы ДИП и ИЭА в прямом варианте, а также в сочетании друг с другом используют для анализа загрязненности воды ионами тяжелых металлов, различными органическими веществами. При этом часто способы пробоподготовки являются гораздо более простыми, чем в спектрометрии или газовой хроматографии. Преимуществом метода ИЭА является (в отличие от других методов, например, атомно-адсорбционной спектрометрии) также способность “отличать” свободные ионы от их связанных химических форм, что важно и для оценки физико-химических свойств анализируемых веществ, и с точки зрения биологического контроля (например, при оценке токсичности вод). Время проведения анализа иногда сокращается до нескольких секунд за счет повышения скорости развертки поляризующего напряжения.
Потенциометрия
с применением различных ионоселективных электродов используется в анализе воды для определения большого числа неорганических катионов и анионов. Концентрации, которые удается определить таким способом, 100
-10-7
моль/л. Контроль с помощью ионоселективных электродов отличается простотой, экспрессностью и возможностью проведения непрерывных измерений. В настоящее время созданы ионоселективные электроды, чувствительные к некоторым органическим веществам (например, алкалоидам), поверхностно-активным веществами и моющим веществам (детергентам). В анализе воды используются компактные анализаторы типа зондов с применением современных ионоселективных электродов. При этом в ручке зонда смонтирована схема, обрабатывающая отклик, и дисплей.
Кондуктометрия
используется в работе анализаторов детергентов в сточных водах, при определении концентраций синтетических удобрений в оросительных системах, при оценке качества питьевой воды. В дополнение к прямой кондуктометрии для определения некоторых видов загрязнителей могут быть использованы косвенные методы, в которых определяемые вещества взаимодействуют перед измерением со специально подобранными реагентами и регистрируемое изменение электропроводности вызывается только присутствием соответствующих продуктов реакции. Кроме классических вариантов кондуктометрии применяют и ее высокочастотный вариант (осциллометрию), в котором индикаторная электродная система реализуется в кондуктометрических анализаторах непрерывного действия. [15, 8-11]
2.1 Аппаратура для хроматографии
Газоанализаторы.
1. Универсальный газоанализатор «ГАНК-4».
ГАНК-4 – серийная копия Первого универсального космического газоанализатора.
ГАНК-4 отмечен высшими наградами на 5 международных выставках, имеет 7 патентов.
В газоанализаторе реализован экспрессный метод измерения максимально-разовых концентраций основных загрязнителей атмосферного воздуха и воздуха рабочей зоны (контролируемые вещества по выбору – до 130 (аммиак. ацетон, аэрозоль краски, бензин, водород, диоксид азота, диоксид углерода, кислород, кислота азотная, серная, уксусная, ксилол, марганец, масло минеральное, метанол, озон, окись этилена, пыль, сажа, сероводород, стирол, толуол, фенол, формальдегид. щелочь, этанол. этиленгликоль. этилцеллозольв и другие); диапазон измерений – от 0,001 мг/куб. м до 100 % об).
Достоинством газоанализатора является малый вес, автономное питание от встроенного аккумулятора, возможность легкого и быстрого использования различных датчиков (химических сенсоров) и химкассет на различные ингредиенты для анализа атмосферного воздуха и воздуха рабочей зоны. Он с успехом применяется как основное средство измерений при аттестации рабочих мест.
Широкая номенклатура анализируемых веществ дает возможность пользователю легко выбирать перечень загрязнителей и диапазоны их измерения, которые необходимы для работы на различных объектах.
Газоанализатор «ГАНК-4» удобен в эксплуатации, не требуется специальной перенастройки прибора при переходе с одного ингредиента на другой, имеется возможность производить измерения в любом месте, на любой высоте, не требуется выполнять усредненный расчет показаний – средний результат высвечивается на дисплее, одновременно производится отбор следующей пробы и обработка результата.
Газоанализатор является незаменимым средством измерения при различной загазованности объектов, ликвидации чрезвычайных ситуаций, для принятия срочных мер.
2. Портативный газовый хроматограф ФГХ-1.
ФГХ-1 является современным автоматизированным средством экспресс определения концентрации вредных веществ в воздухе. Благодаря высокой чувствительности и автоматизации, один и тот же прибор без какой-либо пробоподготовки позволяет анализировать содержание вредных веществ в воздухе в широком диапазоне концентраций: от ПДК в атмосфере до промышленных выбросов и при чрезвычайных ситуациях.
ФГХ - является уникальным средством эксресс-анализа, предназначенным для работы как в лабораторных, так и в «полевых» условиях непосредственно на исследуемом объекте, так как содержит собственные средства электро- и газового питания. Результаты анализа, комментарии к ним и сами хроматограммы автоматически документируются в памяти компьютера и могут быть немедленно предъявлены Заказчику или администрации контролируемого предприятия. Количество хранящихся хроматограмм – до 2000. Помимо определения концентрации веществ, ФГХ позволяет их автоматически идентифицировать.
Хроматограф содержит компьютер типа «Note-Book». Простое в использовании программное обеспечение позволяет проводить анализ в автоматическом режиме, а также работать с хроматограммой, воспроизводимой на экране компьютера. Для работы на ФГХ в автоматическом режиме не требуются специальные знания и опыт работы на хроматографах.
Анализируемые вещества – предельные и непредельные углеводороды, спирты, простые и сложные эфиры, ароматические углеводороды, кетоны, нефтепродукты, растворители, хлорпроизводные углеводородов, окись азота, сероуглерод и другие. Время анализа – менее 10 минут.
Некоторые примеры наиболее подходящих портативных (переносных) средств и их основные характеристики:
1. Хроматограф газовый полевой типа ЭХО-М (г. Новосибирск) масса 6 - 7 кг, электропитание 12 В, время непрерывной работы 8 ч. Детектор электронного захвата. Возможна замена детекторов (фотоионизационный детектор, пламенно-ионизационный детектор). Предел обнаружения с детектором электронного захвата составляет 5 10-13 кг (с возможным дополнением 1000 - кратного обогащения в выносном концентраторе). Цена - 12 000 - 14 000 $.
2. Хроматограф газовый переносной для анализа неорганических газов и продуктов сгорания топлива типа АХГ - 002. Предел обнаружения, г/см3: по Н2
- 8,4·10-10
, по СО - 3,5·10-8
, по СН4
- 6,6·10-9
, по О2
- 8,7·10-9
, по СО2
- 9,2·10-7
с детектором по теплопроводности. Цена ~ 2100 $.
3. Хроматограф газовый малогабаритный типа ХПМ - 5 для анализа сложных смесей веществ. Масса - 20 кг (аналитический блок) и 8 кг (блок питания), габариты, мм - 412х282х341 (аналитический блок) и 120х311х290 (блок питания). Пределы обнаружения: S - 1,0·10-10
(пламенно-фотометрический детектор), P - 1·10-11
- (пламенно-фотометрический детектор) и 2,0·10-12
(термоионный детектор), N-5·10-12
(термоионный детектор), пестициды - 4,0·10-13
(детектор электронного захвата), УВ - 2,0·10-8
(детектор по теплопроводности) и 2·10-11
(пламенно-ионизационный детектор). Цена 3500 $.
4. Хроматографы жидкостные переносные типа «Цвет - 403». Масса - 16 кг, предел обнаружения, в мг/мл: 10-8
- 10-10
(электрохимический детектор) и 10-4
(ультрафиолетовый детектор). Цена 3000 - 3400 $.
5. Фотометр КФК-05 переносной малогабаритный (АООТ «Загорский оптико - механический завод», г. Сергиев-Посад). Габариты 190х170х83 мм, вес 1,2 кг, электропитание 220 и 12 В. Погрешность 1 %, среднеквадратичное отклонение 0,15 %.
6. Микрофотоколориметр полевой. МКМФ-02П (микропроцессорный аналог). Цена 455 - 520 $.
7. Спектрофотометр переносной DR/2010 VIS, =400-900 нм, погрешность 2 %, среднеквадратичное отклонение 0,15 %. Цена 3500 $.
Хроматографы
Вторым признанным лидером по числу реализуемых методик анализа веществ в объектах окружающей среды (20 - 40 %) в настоящее время являются приборы, основанные на хроматографии. Газовые (подвижная фаза - газ, неподвижная - твердый сорбент), газожидкостные (подвижная фаза - газ, неподвижная - тонкий слой жидкости на твердом носителе), жидкостные (подвижная фаза - жидкость, неподвижная фаза - твердый сорбент).
Среди отечественных хроматографических приборов больше всего отмечается газовых хроматографов (ряд серий и несколько десятков моделей). Наиболее известными в России являются газовые хроматографы серии «ЦВЕТ» Дзержинского завода (Московская область). Наиболее распространенная модель из этой серии - лабораторный газовый хроматограф «ЦВЕТ-800» с пламенно-ионизационным детектором. Цена базовой модели от 3700 $. Она может комплектоваться еще пятью детекторами (290 - 860 $).
ДТП - детектор по теплопроводности (для анализа летучих органических и неорганических соединений), неселективен,
ДЭЗ (ЭЗД) - детектор электронного захвата. Для высокочувствительного анализа Cl-, P- и N- содержащих соединений, в том числе ядохимикатов, селективен к Cl и O содержащим соединениям,
ПФД -пламенно - фотометрический детектор, селективен к P- и S-содержащим соединениям,
ТИД - термоионный детектор, селективный к P- и N- содержащим соединениям,
ФИД - фотоионизационный детектор (для анализа ароматических и алифатических углеводородов, фенолов, пестицидов и др. органических веществ с потенциалом ионизации ниже 12 эВ).
В зависимости от детектора и определяемого вещества чувствительность этого хроматографа может составлять 10-10
- 10-4
% об. Отличается высокой точностью (± 1-7 %) и воспроизводимостью анализа. Режимы задаются и управляются микропроцессором, а обработка выходной информации осуществляется компьютером или с выводом на самописец для ручной обработки.
Еще одна достаточно известная модель газовых хроматографов - «Кристалл». Наиболее современные и полностью автоматизированные отечественные лабораторные хроматографы - «Кристалл-200М», «Кристалл-4000».
Жидкостные хроматографы
Наиболее известны отечественные микроколоночные лабораторные жидкостные хромтаографы серии «МИЛИХРОМ», управляемые компьютером (5400 - 8400 $). Эти приборы позволяют с чувствительностью 10-9
- 10-11 г (10-3
- 10-5 г в пробе) определять пестициды, фенолы, тяжелые металлы, ПАУ, альдегиды, бензойную кислоту и другие органические вещества. Точность определения обычно составляет 1 - 3 %.
Отечественные ионные хроматографы: «ЦВЕТ - 3006М», «ЦВЕТ - 4000», «Стайер».
Остановимся более подробно на принципе работы детекторов, используемых в хромтаографии.
Детекторы газовых хроматографов
Детекторы обычно классифицируют на основании их селективности на универсальные, реагирующие на каждый компонент в подвижной фазе, селективные для определенной группы веществ, специфические для одного или ограниченного круга компонентов со сходными химическими характеристиками.
Пламенно-ионизационный детектор (ПИД). Проводимость газа -носителя, являющегося электрополяризатором, существенно возрастает благодаря ионам, образующимся при горении органических соединений в водородном пламени. Отклик ПИД пропорционален числу атомов углерода в молекуле, изменяется при переходе от одного класса органического соединений к другому незначительно.
Достоинства
: простота в обращении, быстрый отклик, широкий линейный динамический диапазон, универсальность.
Недостатки
: при проведении анализа определенного соединения в сложной матрице требуется более селективный детектор для уменьшения числа пиков мешающих компонентов. ПИД дает слабый отклик на вещества с малым содержанием углерода.
Электронно-захватный детектор (ЭЗД) используют для определения галогенсодержаищх соединений: хлорорганические пестициды, дибензафураны, тригалометаны и т.д. Принцип действия этого детектора основан на уменьшении проводимости, вызываемом захватом электронов специфическим анализируемым веществом. В состав детектора входит радиоактивный источник малой интенсивности (фольга с 63
Ni), который испускает электроны высокой энергии. Ионизация молекул газа - носителя (азота или смеси аргона и метана) приводит к образованию ионов и тепловых электронов, которые и формируют электрический ток в ионизационной камере. Когда в нее попадают молекулы галогенсодержащих органических соединений, тепловые электроны захватываются атомами галогена и проводимость уменьшается, что приводит к формированию сигнала детектора.
ЭЗД хорошо зарекомендовал себя при анализе питьевых и подземных вод. В случае поверхностных и сточных вод, содержащих множество органических соединений различных классов, требуется предварительная очистка вод.
Сочетание фотоионизационного детектора и детектора электролитической проводимости. Для анализа летучих ароматических и галогенсодержащих соединений рекомендуется последовательное соединение неразрушающего фотоионизационного детектора (ФИД) и детектора по электролитической проводимости (ЭПД).
В фотоионизационном детекторе вещества возбуждаются фотонами, излучаемыми УФ-лампой, электрический ток, формируемый заряженными частицами, измеряется с помощью двух электродов. Селективность зависти от используемой лампы.
При детектировании галогенсодержащих компонентов посредством ЭПД входящее из колонок вещество восстанавливается водородом в никелевой реакционной трубке при 85 о
С с образованием газообразного галогенводорода, который в свою очередь растворяется в н-пропаноле. Изменение проводимости растворителя преобразуется в сигнал детектора.
Атомно-эмиссионный детектор. АЭД позволяет различать галогенорганические соединения. В АЭД выходящее из колонки вещество атомизируется в высокоэнергетическом источнике, образовавшиеся возбужденные атомы излучают свет при возвращении в основное состояние. Излучаемый свет с различными длинами волн диспергируется в спектрометре и измеряется посредством фотодиодной матрицы. Каждый химический элемент имеет свой собственный типичный эмиссионный спектр, в котором эмиссионные линии обычно образуют кластеры с постоянным соотношением интенсивностей внутри кластера.
Комбинированные методы дают дополняющую друг друга информацию, позволяющую произвести правильную идентификации веществ, которые не могут быть опознаны с помощью какого- либо одного метода.[11-12]
Глава 3. Примеры применения хроматографии в анализе объектов окружающей среды
Анализ состояния водной среды с помощью метода газовой хроматографии[13-15]
Метод газовой хроматографии для анализа состояния водной среды все шире проникает из области научного эксперимента в сферы, непосредственно связанные со многими сторонами жизни человека.
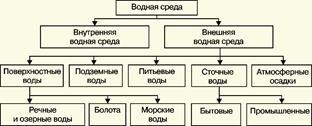
В большинстве случаев термин "водная среда" относят к различным водным объектам, находящимся вне организма человека - к морским и речным водам, иным поверхностным водам суши, подземным водам, промышленным и бытовым стокам, атмосферным осадкам, наконец, к пищевым водам (схема 1). Все эти группы объектов было бы правильнее называть "внешней водной средой".
Схема 1
В связи с тем, что в организме человека, как и многих других живых существ, по крайней мере 80% приходится на долю воды, правомерно говорить о "внутренней водной среде" и о соответствующих объектах анализа. Эта категория объектов включает гомогенизаты и экстракты тканей различных органов и биологические жидкости, к числу которых относятся плазма крови, лимфа, слюна, моча, желчь, желудочный сок, спинно-мозговая жидкость и другие (схема 2). Как в первой, так и во второй группе объектов в анализируемых пробах могут присутствовать весьма разнообразные вещества, детальный анализ которых требует применения различных аналитических методов. Так, например, растворенные газы в морских и подземных водах, а также и в крови человека, определяют с помощью спектральных методов и в ряде случаев с помощью газовой хроматографии. Металлы, присутствующие в водных объектах, анализируют с помощью атомной абсорбционной спектроскопии или эмиссионной спектроскопии с индуктивно связанной плазмой. Очень малые следовые примеси многих элементов в водной среде определяют с помощью радиоактивационного анализа, а состав присутствующих в водной среде анионных составляющих анализируют методом ионной хроматографии.
Схема 2

Высокомолекулярные полимерные вещества, присутствующие в водной пробе (гуминовые и фульвиновые кислоты во внешних природных водах, белковые компоненты и нуклеиновые кислоты в плазме крови), определяют методами жидкостной хроматографии - эксклюзионной, тонкослойной и высокоэффективной жидкостной хроматографии (схема 3).
Схема 3

Однако очень большое число низкомолекулярных летучих органических соединений, способных переходить в парообразное состояние без разложения, анализируют и определяют количественно с помощью метода газовой хроматографии (схема 4). При этом собственно газохроматографическому определению может предшествовать операция извлечения анализируемых компонентов из водной пробы, их концентрирование и в ряде случаев перевод в их производные, обладающие более высокой летучестью либо меньшей полярностью, чем исходные соединения, и потому более пригодные для анализа с помощью газовой хроматографии (например, органические кислоты обычно переводят в их метиловые эфиры, аминокислоты - в алкиловые эфиры N-трифторацетильных производных и т.п.). Таким образом оказывается возможным использовать метод газовой хроматографии для определения тех органических веществ, которые в принципе не переходят в пар без разложения вследствие своей высокой полярности или малой термической устойчивости (например, углеводы).
Схема 4

Метод газовой хроматографии уже в течение достаточно длительного времени является общепризнанным способом анализа летучих органических компонентов и следовых примесей водной среды. Опубликованы многочисленные монографии и оригинальные статьи, описывающие особенности применения метода газовой хроматографии к анализу тех или иных водных объектов.
Как и любой другой аналитический метод, газовая хроматография может дать корректную объективную информацию о составе водных объектов при условии достаточно правильного выполнения предварительных операций по отбору представительных проб анализируемых вод, извлечения и концентрирования подлежащих определению компонентов и их групп и ввода их в хроматографическую систему.
Объектами газохроматографического определения в водных средах могут быть растворенные газы и органические соединения с молекулярной массой от 16-30 (метан, этан) до 400-500 и более. Состав примесей может довольно быстро изменяться вследствие целого ряда причин. Поэтому время от момента отбора проб до выполнения анализа или предшествующих ему операций, обеспечивающих консервацию их состава, должно быть по возможности малым.
Причины изменения состава примесей водной среды, определяемых с помощью газовой хроматографии, могут включать следующие процессы:
а) потери растворенных газов и наиболее легколетучих органических компонентов вследствие изменений температуры и давления (например, при нагреве водной пробы от исходной температуры водоисточника до температуры лабораторного помещения);
б) исчезновение некоторых подлежащих определению примесных компонентов водных проб в результате химических и микробиологических процессов при хранении до проведения анализа (окисление альдегидов и тиолов, гидролиз ацеталей и галогенопроизводных, микробиологическое расщепление углеводородов нефти и др.);
в) загрязнение проб примесями, извлекаемыми из полимерной тары, используемой при отборе проб и их последующей транспортировке и хранении (пластификаторы, низкомолекулярные компоненты полимеров и т.п.);
г) загрязнение проб примесями, содержащимися в применяемых для экстракции растворителях;
д) изменение проб в испарителях и колонках применяемых хроматографических систем.
Правильно организованный газохроматографический анализ должен в возможно более полной степени исключить все перечисленные выше причины изменения состава анализируемых проб. Этой цели служат многочисленные методики анализа, опубликованные в оригинальных работах, а в ряде случаев и включенные в нормативные документы.
Для извлечения определяемых компонентов из водных матриц применяют методы экстракции малыми объемами органических растворителей с последующим концентрированием путем отгонки экстрагента (рис. 1); твердофазной экстракции с сорбцией определяемых компонентов на адсорбентах с привитой органической неподвижной фазой (углеводородными радикалами от С2
H5
до С20
Н41,
либо функционально замещенными фрагментами с нитрильными, аминными или диольными группами). Разработаны методы микроэкстракции на единичном стеклянном или кварцевом волокне, покрытом пленкой полисилоксановой неподвижной фазы.

Рис. 1. Типичные хроматограммы нефтяных загрязнений, экстрагированных из воды Таганрогского залива: а) - август 1991 г.; б) - октябрь 1991 г.
В пробу анализируемой воды объемом около 100 мл (в конической колбе) помещают магнитную мешалку в форме стеклянной трубки длиной 30 мм и диаметром 2-3 мм с запаянными концами. Внутрь трубки помещают стальной стержень длиной 25 мм и диаметром 1-1,5 мм, а снаружи на трубку надевают отрезок трубки из силиконовой резины длиной 25-30 мм с внутренним диаметром 1 мм и толщиной стенок 0,5 мм. Согласно опубликованным данным перемешивание водной пробы такой мешалкой со скоростью несколько сотен оборотов в минуту в течение 10-15 мин при 20 °С приводит к тому, что более 90% всех липофильных примесей абсорбируется в силиконовой оболочке мешалки. После этого мешалку можно поместить в нагретый испаритель хроматографа для немедленного проведения анализа либо сохранить ее длительное время в закрытой пробирке для транспортировки в стационарную лабораторию.
Подобная техника извлечения и концентрирования малых примесей, названная авторами Stir Bar Sorptive Extraction, несомненно, имеет серьезные перспективы широкого применения. Возможно, что использование таких магнитных мешалок с полимерными покрытиями разных типов позволит избирательно извлекать из водных проб различные группы соединений, отличающиеся по своей полярности и прочим физико-химическим характеристикам.
Для определения состава легколетучих компонентов водных проб оказывается плодотворным метод газохроматографического анализа равновесного пара и газовой экстракции с улавливанием извлекаемых веществ в сорбционных концентраторах и последующим криогенным вводом в капиллярную колонку. Таким способом, например, подробно изучен состав хлорсодержащих микропримесей, образующихся при хлорировании питьевой воды (рис. 2). При извлечении микропримесей полярных веществ, хорошо растворимых в воде, применяют метод экстракции полярными водорастворимыми экстрагентами (спиртом, ацетоном) с предварительным насыщением водных проб неорганическими солями (высаливание хлоридом натрия или сульфатом аммония).
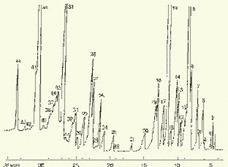
Рис. 2. Хроматограмма летучих органических соединений, содержащихся в пробе хлорированной питьевой воды. Капиллярная колонка длиной 50 м и диаметром 0,25 мм с силиконовой смазкой "Эдвардс" в качестве неподвижной фазы. Пики, обозначенные цифрами, идентифицированы. Пик 9 – хлороформ
Собственно газохроматографический анализ в большинстве случаев в настоящее время осуществляют с применением высокоэффективных капиллярных колонок в условиях программирования температуры от 50-100 °С до 300-350 °С (и даже до 400 °С и выше). Скорость нагрева колонок при этом может изменяться от 3-5 до 20-30 град/мин в зависимости от характера разделяемых компонентов.
Идентификацию пиков на хроматограммах выполняют с применением известных хроматографических зависимостей индексов удерживания от температур кипения и от молекулярной массы веществ. Для той же цели применяют селективные детекторы (например, электронно-захватный), термоионные хемилюминесцентные, атомно-абсорбционные, масс-селективные и др.
Наиболее полную информацию о молекулярном строении определяемых веществ позволяют получить методы хромато-масс-спектрометрии и газовой хроматографии с ИК-Фурье спектроскопическим детектором. Особенно плодотворным оказалось использование этих двух методов при определении пестицидов, полихлорированных бифенилов, диоксинов и им подобных веществ, продуктов распада токсичных ракетных топлив и боевых отравляющих веществ в объектах окружающей среды (рис. 3).
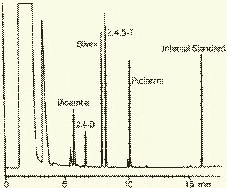
Рис. 3. Хроматограмма воды реки Оук Крик (США), содержащей пестициды: 1 - Дикамба (4,7 мкг/л); 2 - 2,4-D (7,0 мкг/л); 3 - Силвекс (4,5 мкг/л); 4 - 2,4,5-Т (5,2 мкг/л); 5 - Пиклорам (3,3 мкг/л); 6 - внутренний стандарт
В ряде случаев применение селективных детектирующих систем позволяет не только повысить чувствительность определения целевых компонентов, но и выявить источники загрязнения водной среды. Так, например, применение хемилюминесцентного озонового детектора с высокой специфической чувствительностью к веществам, содержащим серу, позволяет зафиксировать профиль серосодержащих соединений нефти и нефтяных топлив. Эти компоненты значительно более устойчивы в водной среде, чем углеводородные составляющие нефтепродуктов, поэтому совпадение таких профилей позволяет с большой степенью достоверности указать источник нефтяного загрязнения данного водного бассейна (рис. 4), .

Рис. 4. Хроматограммы: а) - серосодержащих компонентов нефтяного загрязнения морской воды (район г. Таганрога); б) - судового дизельного топлива.
Кварцевая капиллярная колонка длиной 25 м и диаметром 0,25 мм с полидиметилсилоксаном SE-54 в качестве неподвижной фазы. Программирование температуры от 60 до 280 °C cо cкоростью нагрева 10 град/мин: 1 - метилбензотиофены; 2 - диметилбензотиофены; 3 - дибензотиофен; 4 - метилдибензотиофены; 5 – диметилдибензотиофены
Большие возможности газохроматографического анализа в настоящее время определенно позволяют осуществить достаточно полный анализ водных проб практически любого происхождения, в том числе питьевых, речных, озерных и морских вод, сточных вод коммунальных систем и промышленных предприятий.
В ряде случаев такие анализы могут быть проведены в полевых условиях непосредственно в местах отбора проб. Тем не менее, пока не существует методов, позволяющих провести полный анализ водных проб в единой системе без проведения предварительных лабораторных операций, часто трудоемких и длительных.
Перспективными для создания таких методов следует считать хроматографические системы с использованием в качестве подвижных фаз парообразных и сверхкритических сред (в том числе паров воды).
Широко используется газовая хроматография также и для анализа объектов внутренней водной среды. При этом применяется весь арсенал технических приемов, разработанных за 50 лет развития газовой хроматографии: высокоэффективные капиллярные колонки, высокочувствительные селективные детекторы, комбинированные аналитические системы, сочетающие газовую хроматографию с масс-спектрометрией и с ИК-спектроскопией с Фурье-преобразованием.
В этой области сложились два основных направления аналитических исследований. С одной стороны, это изучение состава естественных сред организма (плазмы крови, мочи, слюны, спинно-мозговой жидкости и др.) с целью выявления изменений этого состава в зависимости от физиологических особенностей организма, наличия патологических изменений и тому подобных факторов.
В настоящее время это направление газохроматографического анализа внутренней водной среды организма отражено в ряде руководств и монографий. Показано, что во многих случаях газохроматографический анализ позволяет выявить на ранней стадии целый ряд заболеваний и таким образом ускорить их лечение (рис. 5). При таких исследованиях широко используют экстракцию целевых компонентов растворителями, твердофазную экстракцию и метод анализа равновесного пара (рис. 6).
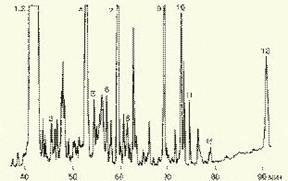
Рис. 5. Метаболический профиль стероидов из мочи пациента с опухолью яичника, секретирующей тестостерон. Идентифицированы все компоненты. Увеличенное содержание компонентов 1, 2, 4 и 7 указывает на наличие опухоли (1 - андростерон; 2 - этиохоланон; 4 - 11b-оксиандростерон; 7 - прегнантриол)

Рис 6. Хроматограмма летучих органических соединений мочи, полученная на капиллярной колонке с полисилоксановой неподвижной фазой БС-2 в условиях программирования температуры: 1 - ацетон; 3 - этанол; 6 - 2-пентанон; 7 - н-пропанол; 10 - диметилдисульфид;13 - 4-гептанон; 14 - н-бутанол; 15 - 2-гептанон; 29 - пиррол; 45 – карвон
Вторым важным направлением в анализе объектов внутренней водной среды является выявление чужеродных для организма соединений (фармацевтических препаратов, разного рода ядов, алкоголя, других наркотических веществ и т.п.). Так, применение высокочувствительного термоаэрозольного детектора, избирательно регистрирующего азотсодержащие соединения, позволило регистрировать противосудорожные лекарственные средства в крови детей, больных эпилепсией, даже через 3-5 дней после их применения (рис. 7). Аналогично, с помощью селективного термоионного детектора с высокой чувствительностью регистрировали в плазме крови наличие анестезирующего препарата кетамина, находящего, к сожалению, довольно широкое применение в качестве галлюциногенного наркотика (рис. 8).

Рис. 7. Хроматограмма 0,1% раствора противоэпилептических лекарственных средств. Кварцевая капиллярная колонка длиной 10 м и диаметром 0,25 мм с полисилоксаном OV-17 в качестве неподвижной фазы: 1 - этанол; 2 - фенобарбитал; 3 - гексамидин; 4- дифенин

Рис. 8. Хроматограмма смеси компонентов плазмы крови, содержащей кетамин: а) - пламенно-ионизационный детектор; б) - термоионный детектор; 1 - кетамин; 2 - внутренний стандарт
Подобным же способом при газохроматографическом анализе проб плазмы крови, мочи или слюны могут быть определены несколько сотен других наркотиков, веществ, используемых в качестве допинга в спортивных соревнованиях, и анаболических стероидов, запрещенных к употреблению международными спортивными организациями.
Все перечисленные выше достижения газохроматографического метода в области анализа объектов водной среды все шире проникают из области научного эксперимента в сферы, непосредственно связанные с многими сторонами жизни современного общества. К таким областям можно отнести изучение состояния окружающей среды, контроль качества питьевой воды, сельскохозяйственной продукции и пищевых продуктов, клиническую медицину, криминалистику и ряд других.
Определение полиароматических углеводородов в объектах окружающей среды методами жидкостной и тонкослойной хроматографии[14]
Было определено содержание полиароматических углеводородов (ПАУ), в частности, бенз(а)пирена в снежном покрове. Пробоподготовку осуществляли экстракцией диэтиловым эфиром. Качественный анализ осуществляли методом тонкослойной хроматографии. Пробы наносили на пластину Silufol UV-254 и осуществляли хроматографический анализ в двух системах: система 1 - раствор кофеина в хлороформе; система 2 - смесь циклогексана и н-гексана. Использование системы 1 позволило снизить нижний предел обнаружения.
Количественный анализ осуществляли методом газожидкостной хроматографии (ГЖХ). Исследование проводили на хроматографе «Цвет-500» с пламенно-ионизационным детектором. В качестве сорбента использовали силиконовый каучук SE-54 с нанесенной на него неподвижной фазой OV-101. Анализ проводили в режиме программирования температуры от 200 до 310 С со скоростью 4 С /мин. В качестве газа-носителя использовали азот. Метод позволил определить ПАУ на уровне ПДК.
Глава 4. Современное аппаратурное оформление

Портативные хроматографы Agilent Micro-GC
Портативные газовые хроматографы (ГХ), занимая существенно меньше места по сравнению с лабораторными газовыми хроматографами, обеспечивают при этом сопоставимое качество анализов. Небольшие, размером с коробку из-под ботинок, Микро газовый хроматограф позволяют анализировать компоненты в концентрациях порядка одна часть на миллион (ррт) за несколько секунд - в десятки раз быстрее, чем обычные лабораторные газовые хроматографы. Микро газовые хроматографы предназначены для анализа газовых смесей или веществ с низкой температурой кипения (до 90 С) непосредственно в цехах, "у реактора". В корпус газового хроматографа с наибольшим размером 46 см помещаются до 4 хроматографических модулей. Микро газовый хроматограф характеризуется высокой производительностью его легко переносить, он быстро приводится в рабочее состояние. Это идеальный хроматограф, для получения быстрых результатов, например, при контролировании процессов в опытных установках, или, когда необходимо убедиться в однородности множества образцов газовых смесей. Использование модема позволяет передавать информацию, полученную от микро газового хроматографа по телефонным линиям на большие расстояния.
Революционное повышение производительности газового хроматографа
Миниатюризация инжекторов и детектора по теплопроводности обусловила использование в микро газовом хроматографе коротких и очень тонких капиллярных колонок, что более повысило эффективность процесса разделения. Это дает возможность проанализировать, например, природный газ (СГС10) за время не более 150 сек, смеси неорганических газов с серосодержащими газами - менее чем за 30 сек, смеси легколетучих соединений -за 50 сек, а сложную смесь нефтезаводских газов -за 160 сек. Наличие миниатюрной схемы разделения с обратной продувкой позволяет микро газовому хроматографу анализировать смеси, содержащие тяжелые компоненты, и быть готовым к началу следующего такого анализа уже через 2-3 минуты. Сокращение времени анализа в десятки раз приводит к революционному скачку в повышении производительности труда, помогает быстрее принимать правильные решения на производстве.

Микро-технология на монокристаллах кремния
Благодаря развитию микротехнологии в производстве газового хроматографа основные его узлы изготавливаются на основе монокристаллического кремния: инжектор с вентилями, система обратной продувки и универсальный детектор по теплопроводности. Эти основные узлы, а также колонка сравнения и рабочая колонка и термостат собраны в один механический прочный высокопроизводительный модуль.
Каждый газовый хроматограф может быть составлен из нескольких (до четырех) независимо управляемых модулей, сочетающих высокую скорость анализа с точностью результатов, требуемых в промышленности. Отсутствие подвижных частей в инжекторе делает всю систему исключительно надежной и долговечной.
Десять компонентов – менее чем за 120 сек!
Литература
1. Жуховицкий А.А., Туркельтауб Н.М. Газовая хроматография. М.: Гостоптехиздат, 1962, 240 с.
2. Сакодынский К.И., Киселев А.В., Иогансен А.В. и др. Физико-химическое применение газовой хроматографии. М.: Химия, 1973. - 254 с.
3. Жидкостная колоночная хроматография. В 3 т. / Под ред. З.Дейла, К.Мацека, Я.Янака. М.: Мир, 1972. – 439с.
4. Березкин В.Г., Алишоев В.Р., Немировская И.Б. Газовая хроматография в химии полимеров. М.: Наука, 1972. - 287 с.
5. Морозов А.А. Хроматография в неорганическом анализе. М.: Высш. шк., 1972. - 233 с.
6. Березкин В.Г., Бочков А.С. Количественная тонкослойная хроматография. М.: Наука, 1980. - 183 с.
7. Лабораторное руководство по хроматографическим и смежным методам. В 2 т. / Под ред. О.Микеш. М.: Мир, 1982, т. 1–2, 783 с.
8. Кирхнер Ю. Тонкослойная хроматография. В 2 т. М.: Мир, 1981, т. 1, 615 с.; т. 2. - 523 с.
9. Экстракционная хроматография. / Под ред. Т.Браун, Г.Герсини. - М.: Мир, 1978. - 627 с.
10. Скуг Д., Уэст Д. Основы аналитической химии / Пер. с англ. В 2 т. М.: Мир, 1979. – 324с.
11. Гольдберг К.А., Вигдергауз М.С. Введение в газовую хроматографию. М.: Химия, 1990. – 278с.
12. Хмельницкий Р.А., Бродский Е.С. Хромато-масс-спектрометрия. М.: Химия, 1983. – 280с.
13. Горелик Д.О., Конопелько Л.А., Панков Э.Д. Экологический мониторинг. В 2 т. СПб.: Крисмас, 2002. – 457с.
14. Назаркина С.Г. Определение полиароматических углеводородов в объектах окружающей среды методами жидкостной и тонкослойной хроматографии.
15. Хроматографический анализ окружающей среды. / Под ред. Р.Гроба. М.: Мир, 1979. - 606 с.
|