Оглавление
I. Введение
II. Перегруппировки карановых монотерпеноидов, протекающие по ионному механизму
III. Перегруппировки, протекающие по радикальному механизму
IV. Согласованные реакции
V. Выводы
I. Введение
Одним из наиболее распространенных монотерпеновых углеводородов является 3-карен — 3,7,7-триметилбицикло[4.1.0]гепт-3-ен (1
), входящий в состав многих эфирных масел и скипидаров.
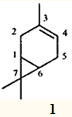 молекулярный радикальный перегруппировка карановый монотерпеноид молекулярный радикальный перегруппировка карановый монотерпеноид
Интерес к 3-карену обусловлен наличием в его молекуле гем-
диметилциклопропанового кольца — уникального фрагмента, являющегося важным структурным элементом ряда биологически активных соединений. Здесь можно упомянуть природные и синтетические пиретроиды, стеркуловую кислоту и ее эфиры, производные аминоциклопропанкарбоновой кислоты и др. Кроме того, 3-карен и родственные соединения, как и другие бициклические терпеноиды, способны к разнообразным скелетным перегруппировкам. Поэтому они — чрезвычайно перспективные модели для исследований в области фундаментальной органической химии, а также полезные исходные соединения для получения ценных продуктов — сесквитерпеноидов,3
феромонов,4
ювеиоидов,5
пиретроидов 6-7
и душистых веществ.7-9
Известные в настоящее время скелетные перегруппировки соединений ряда карана протекают с образованием структур нескольких типов: с бицикло[3.1.0]гексановым, бицикло[3.2.0]гептановым, п
- и м
-ментановым и 1,1,4-триметилциклогептановым скелетами. По механизмам перегруппировки классифицируются на ионные, радикальные и согласованные.
Существующие обзоры по карановым монотерпеноидам были написаны в основном более 25 лет тому назад и не включают скелетные перегруппировки, исследованные в последние десятилетия. Подробный обзор 10
посвящен перегруппировкам, протекающим по ионному механизму. В обзоре 11
в основном приведены перегруппировки карановых монотерпеноидов, в которых циклопропановое кольцо (ЦПК) раскрывается по внешним связям с образованием соединений ряда п
- и м
-ментана.
В обширной монографии Эрмана "Химия монотерпенов" 12
прекрасно изложены систематические данные по реакциям монотерпенов и рассмотрены механизмы этих реакций, однако фактический материал по превращениям терпеноидов ряда карана включает литературу до 1976 г. К настоящему времени получены новые результаты и появились новые представления о механизмах некоторых реакций, рассмотренных в этой монографии. Периодически публикуемые обзоры13-14
носят констатирующий, а не аналитический характер, поэтому механизмы описанных превращений в них как правило не рассматриваются и не обсуждаются. В последних обзорах, посвященных перегруппировкам терпеноидов, 3,15,16
превращения соединений с карановым скелетом практически не рассматриваются.
Целью курсовой работы является обобщение данных по скелетным превращениям терпеноидов каранового ряда. Более ранние работы привлечены в тех случаях, когда представляет интерес история вопроса или появилось новое понимание известных фактов. Перегруппировки рассмотрены в соответствии с классификацией по их механизмам.
II. Перегруппировки карановых монотерпеноидов, протекающие по ионному механизму
Реализация ионного механизма при молекулярных перегруппировках карановых монотерпеноидов обусловлена взаимодействием циклопропанового кольца с электронодефицитным карбениевым центром в α- или β-положении к циклу. Интерес к этому типу перегруппировок связан с возможностью образования интермедиатов с делокализованным зарядом. Согласно предложенной в обзоре10
классификации, ионные перегруппировки карановых структур подразделяются на гомоаллильную перегруппировку, перегруппировку с трансаннулярным участием ЦПК и перегруппировку типа Вагнера-Меервейна с сужением шестичленного цикла. Однако эта классификация не отражает в полном объеме разнообразие ионных перегруппировок карановых структур и является условной. Гомоаллильная перегруппировка — не единственная возможность скелетных превращений через карбокатион с делокализованным зарядом: в карановых структурах, содержащих двойные связи, может существовать и ион типа пентадиенильного, причем пути его образования могут быть различными для различных соединений. Кроме того, по нашему мнению, перегруппировку с трансаннулярным участием ЦПК и перегруппировку типа Вагнера - Меервейна более корректно рассматривать в ряду согласованных реакций, поскольку в них происходят алкильные сдвиги в заряженных системах.
1. Карбений-ионные перегруппировки
Молекула 3-карена содержит два реакционноспособных фрагмента: двойную связь и циклопропановое кольцо. Оба эти фрагмента характеризуются повышенной электронной плотностью, что делает молекулу реакционноспособной по отношению к электрофильным агентам. Хорошо известна перегруппировка 3-карена под действием кислот с образованием соединений ряда п
- и м
-ментана.12
Очевидно, что в этом случае циклопропановое кольцо выступает как изолированный реакционный центр, образуя карбениевые ионы 2
или 3
. Двойная связь удалена и не взаимодействует с атомом углерода, несущим положительный заряд. Ионы стабилизируются путем выброса протона или захвата нуклеофила (схема 1
).
Описанное превращение осуществляется в том случае, когда вначале протонируется ЦПК. Очевидно, что присоединение протона к двойной связи углеводорода 1
должно приводить к третичному иону 4
.
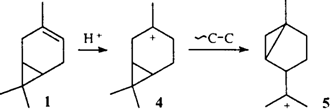
В ионах такого типа возможна перегруппировка, обусловленная так называемым "трансаннуляриым циклопропильным участием"10
(см. раздел IV
.4), но ее результатом было бы образование иона 5
и продуктов иной структуры.
Причину того, что в случае 3-карена протежируется ЦПК, а не двойная связь, можно объяснить с позиций принципа жестких и мягких кислот и оснований (ЖМКО). Циклопропан — более жесткое основание, чем двойная связь (потенциалы ионизации 10.9 и 9.3 эВ соответственно). В молекуле 3-карена разница энергий высших занятых молекулярных орбиталей (ВЗМО) соответствующих фрагментов несколько меньше, но все еще достаточно велика (9.16 и 8.61 эВ).17
Жесткая кислота — протон — присоединяется вначале к жесткому основанию — ЦПК. Так, при взаимодействии эквимолярных количеств 3-карена и хлористого водорода образуются производные со скелетом п
- и м
-ментана.18
Сходство электронного строения двойной связи и ЦПК 19
обусловливает сходство их реакционной способности.20
Наличие катионоидного центра в α-положении к циклопропановому кольцу приводит к их сопряжению и образованию гомоаллильного иона. Направление реакции этого иона с нуклеофилом зависит от пространственных факторов, природы уходящей группы, степени сольватации молекулы (в реакциях сольволиза) и др.

X-
— нуклеофил
Если атака гомоаллильного иона происходит со стороны ЦПК, это приводит к его раскрытию и в случае карановых производных — к перегруппировкам скелета.
Так, в условиях кислотного катализа каран-5-ол (6
) и каран-2-ол (7
) изомеризуются в м
- (8
) и п
-ментен-8-олы (9
) соответственно в результате атаки гидроксила по атому С(8) гомоаллильных ионов 10
и 11
.21,22
Эти реакции можно рассматривать как региоспецифичные.
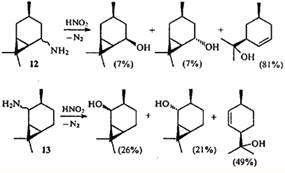
Подобным же образом протекает дезаминирование 5- (12
)- и 2-аминокаранов (13
), однако присутствие среди продуктов бициклических спиртов показывает, что в данном случае реакция не была доведена до конца.23
Гомоаллильные перегруппировки могут сочетаться с другими ионными перегруппировками. Так, при конденсации 3-карена с формальдегидом по Принсу среди продуктов реакции обнаружены два циклических эфира 14
и 15
. Их образование объясняется последовательным протеканием перегруппировки типа Вагнера — Меервейна и гомоаллильной.24-25
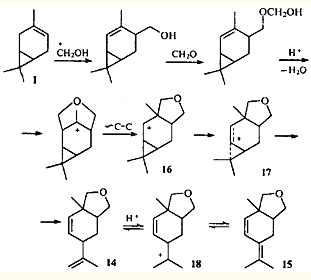
В данном случае перегруппировка Вагнера-Меервейна, протекающая как алкильный сдвиг, не приводит к изменению карановой структуры. Однако алкильный сдвиг возможен лишь потому, что в образующемся вторичном ионе 16
делокализация заряда с участием циклопропанового кольца приводит к его раскрытию и образованию гомоаллильного иона 17
, обладающего меньшей энергией. Ион 17
стабилизируется путем выброса протона от одной из геминальных метильных групп, давая эфир 14
. В кислой среде этот эфир изомеризуется в эфир 15
через классический катион 18
.
2. Перегруппировки, протекающие через ионы с высокой степенью делокализации заряда
Очевидно, что в систему ЦПК - заряженный атом углерода может включаться одна или несколько двойных связей. Образующийся при этом гомопентадиенильный ион характеризуется большей степенью делокализации заряда, чем гомоаллильный, и имеет, соответственно, более низкую энергию. Поэтому реакции с участием гомопентадиенильных ионов протекают легче, чем с участием гомоаллильных. Изомеризация 4-карен-3-ола (19
) в гидроксидиен 20
происходит при кипячении в воде даже в отсутствие кислотных катализаторов:26
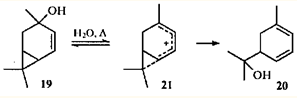
Как и при изомеризации соединений 6
и 7
, необратимость атаки по циклопропановому фрагменту иона 21
обусловливает региоспецифичность реакции.
Следует отметить, что соседство (и, возможно, сопряжение) двойной связи и ЦПК в нейтральной молекуле не является обязательным условием образования гомопентадиенильного иона и, следовательно, протекание скелетной перегруппировки. С точки зрения механизма интересна селективная изомеризация кар-3-ен-5-ола (22
) в м
-мента-1,3-диен-8-ол (20
): двойную связь и ЦПК объединяет в систему сопряжения возникающий между ними карбокатаонный центр.27
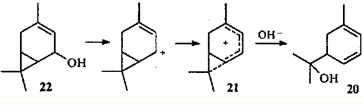
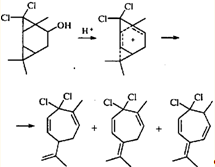
В систему сопряжения катионоидный центр - ЦПК может включаться не только соседняя двойная связь, но также и второй трехчленный карбоцикл. Участие такого интермедиата в реакции приводит к перегруппировке трициклооктанового скелета в замещенный циклогептадиен.28
Эта перегруппировка интересна тем, что в данном случае гем
-дихлорциклопропановое кольцо раскрывается по внутренней связи, а гем
-диметилциклопропановое — по внешней.
Из приведенных выше примеров ионных перегруппировок видно, что для гем
-диметилциклопропанового кольца раскрытие по внешней связи является закономерностью, практически не знающей исключений. Трехчленный цикл, связанный с двумя атомами галогена, напротив, может вести себя по-разному. При метанолизе аддукта 3-карена с дибромкарбеном ЦПК раскрывается по двум направлениям, приводя к продуктам как с сохранением карановой структуры, так и к продуктам перегруппировки.29
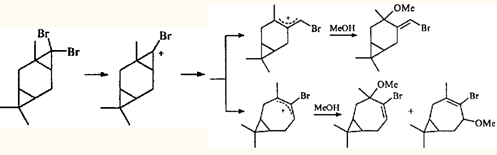
То, что трехчленные циклы в ионных реакциях раскрываются по разным связям, может быть обусловлено природой геминальных заместителей. гем
-Диметильная группировка укорачивает (и усиливает) противолежащую связь циклопропана,30
а два геминальных атома хлора удлиняют и ослабляют ее. Геминальные атомы брома действуют таким же образом, но слабее.31
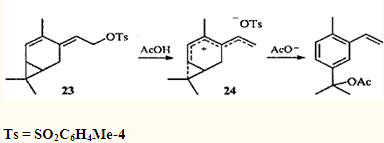
Чрезвычайно легко протекает ацетолиз тозилата 23;
32
в данном случае интермедиатом является гомогептатриенильный ион 24
, в котором степень делокализации заряда еще выше, чем в гомопентадиенильном.
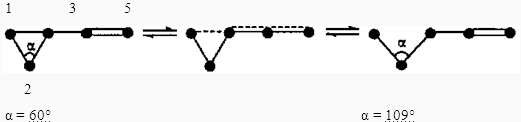
Выше было сказано, что гомоаллильные и гомопентадиенильные перегруппировки являются необратимыми, однако это утверждение не совсем корректно. Действительно, подобные превращения карановых производных можно провести со 100%-ной степенью конверсии. Это обусловлено геометрией иона и образующейся молекулы. Атом углерода ЦПК при С(2), не включенный в цепь сопряжения, в гомоаллильном ионе, в процессе его стабилизации меняет гибридизацию с sp2
на sp3
При этом угол С(1)—С(2)—С(3), равный в молекуле циклопропана 60°, в конечной молекуле увеличивается до 109°, что вызывает, в свою очередь, увеличение расстояния между атомами С(1) и С(3). Однако если эти атомы удастся каким-то образом сблизить, вновь становится возможным образование гомоаллильного иона.
На практике соединения с ментановым скелетом никогда не превращаются в карановые. Однако именно в карановых структурах, благодаря особенностям их строения, можно наблюдать ретро-гомоаллильную перегруппировку, при которой гомоаллильный ион образуется из линейного фрагмента молекулы и приводит к замыканию трехчленного цикла. Эффект подобного рода наблюдался при взаимодействии тозилата 25
с лчтийалюминийгидридом,33
триэтилалюминием,34
а также при его сольволизе.35
Очевидно, что в идеальном случае эта реакция должна протекать по механизму SN
1. Однако скорее всего в реакции участвуют контактные ионные пары: происходит очень сильная поляризация субстрата с почти полным разделением зарядов, но образовавшийся катион стабилизирован противоионом. Отщепление тозилатной группы в соединении 25
приводит к катиону с зарядом на первичном атоме углерода, который гораздо менее стабилен, чем третичные ионы, образующиеся при протонировании ЦПК. Стабилизация иона может осуществляться путем сопряжения заряженного атома с двойной связью, причем перекрывание p
-орбиталей происходит не вдоль σ
-связи, а через пространство ("throughspace").

Сближению этих фрагментов способствует тесное сольватное окружение, поскольку процесс должен происходить быстро. Этот путь приводит к образованию гомоаллильного иона 26
и/или дигомопентадиенильного иона 27
. В том случае, когда интермедиатом является ион 27
, происходит совершенно необычная перегруппировка: фактически одна молекула претерпевает одновременно две перегруппировки различной направленности — гомоаллильную, приводящую к раскрытию циклопропанового кольца, и ретро-гомоаллильную, приводящую к замыканию нового трехчленного карбоцикла.
Тут уместно привести чрезвычайно интересный факт: 2-тозилоксиметил-3-карен (28
) при ацетолизе дает тот же эфир 29
, что и 4-тозилоксиметил-2-карен (25
).36
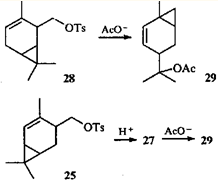
Механизм образования ацетата 29
пока не выяснен. Обращает внимание также отсутствие продуктов, образования которых можно было бы ожидать, опираясь на данные по превращениям эфира 25
. Известно еще одно достаточно необычное превращение тозилата 28
: при пиролизе он дает тот же углеводород, что и тозилат 25
, а именно 1-метил-4-изопропенилбицикло[4.1.0]гепт-2-ен (30
).36
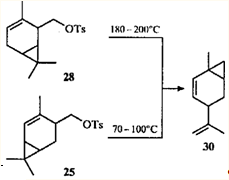
Механизм пиролиза эфира 25
37
будет рассмотрен ниже, как и возможный механизм пиролиза тозилата 28
.
Механизмы ионных перегруппировок, включающих ионы с делокализованным по цепи сопряжения зарядом, являются, конечно, гипотетическими, основанными на строении конечных продуктов. До сих пор неясно, действительно ли возможно сопряжение в таких системах. Ниже, при рассмотрении гидрирования 2-карена (31
) будет показано, что соседство двойной связи и ЦПК отнюдь не гарантирует их сопряжения, по крайне мере в основном состоянии нейтральной молекулы. 17,38
Было бы чрезвычайно полезно проверить с помощью современных квантово-химических расчетов, возможна ли при перегруппировках соединений ряда карана делокализация заряда в системе сопряжения, в которой одним из элементов является ЦПК.
III. Перегруппировки, протекающие по радикальному механизму
Радикальные перегруппировки монотерпеноидов каранового ряда происходят, как правило, при фотохимических реакциях. Фотохимические превращения таких соединений достаточно подробно описаны в обзоре 12
. Для 3-карена перегруппировки в условиях облучения не наблюдались. Известно лишь его фотохимическое окисление с образованием трех гидропероксидов.39
Изомерный 2-карен при облучении в присутствии сенсибилизатора перегруппировывается в 1,4,4-триметилбицикло[3.2.0]гепт-2-ен (32
), который был использован для синтеза грандизола.4
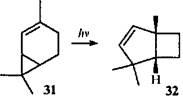
В данном случае механизм перегруппировки не рассматривался, но в литературе описан механизм для родственного соединения — 2-норкарена (33
).40
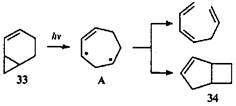
При облучении соединения 33
основным продуктом был цис
-гепта-1,3,6-триен, т.е. преобладал [2π+2ω+2σ]-процесс. Образование небольшого количества бицикло[3.2.0]гепт-2-ена (34
) позволило авторам предложить общий дирадикальный интермедиат А
с семичленным циклом:
Существует точка зрения,12,41
что образование соединений с бицикло[3.2.0]гептановым скелетом при облучении некоторых карановых терпеноидов может протекать как фотоиндуцированная сигматропная перегруппировка. В пользу этого предположения говорит и факт параллельного образования борненовой структуры из кетона 35
.
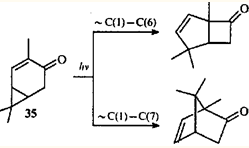
Косвенным подтверждением согласованного механизма перегруппировки кетона 35
можно считать результаты облучения 4-гидроксиметил-2-карена (36
) ртутной лампой высокого давления:42
в присутствии сенсибилизатора (sens) образуются только бициклические стереоизомерные спирты 37
и 38
, а при прямом облучении в гексане — еще и моноциклический спирт 39
.
Кропп, исследовавший эту реакцию,42
проводил аналогию с 1,3-сопряженной системой, постулируя образование аллил-радикалов. При этом ЦПК может претерпевать гомолитический разрыв как "внутренней", так и одной из "внешних" связей. По-видимому, применение сенсибилизатора приводит к тому, что молекула превращается в дирадикал 40
из триплетного состояния: при этом рвется самая слабая связь ЦПК. В синглетном возбужденном состоянии молекула обладает большей энергией, что и приводит к разрыву обеих возможных связей ЦПК. В данном случае происходит разрыв связи С(1)—С(7), а не ее сдвиг. Очевидно, рекомбинация и образование новой связи между атомами С(1) и С(8) радикала 41
(что приводило бы к борнановому скелету) невозможны из-за большего расстояния между этими атомами, чем расстояние между атомами с неспаренными электронами дирадикала 40
.

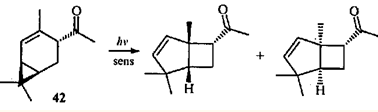
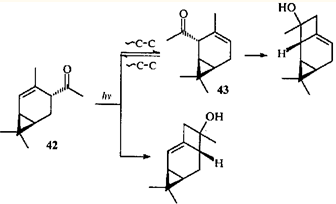
Облучение 4-ацетил-2-карена (42
) в присутствии сенсибилизатора дает продукты с бицикло[3.2.0]гептановой структурой,43
т.е. замена гидроксиметильной группы на ацильную не влияет на механизм реакции.
Однако облучение кетона 42
в отсутствие сенсибилизатора вызывает принципиально иную перегруппировку: образуется продукт 1,3-ацильного сдвига (2-ацетил-3-карен (43
)), и трициклические спирты — продукты циклизации II типа по Норришу обоих присутствующих в реакционной среде кетонов.43
Не совсем обычно ведут себя при облучении цис
- (44
) и транс
-4-караноны (45
). Хотя строение молекул позволяет осуществить расщепление II типа по Норришу связей С(3)—С(4) и С(4)—С(5), среди продуктов фотопревращений цис
-каранона (44
) обнаружены только соединения, образовавшиеся в результате расщепления связи С(4)—С(5).44
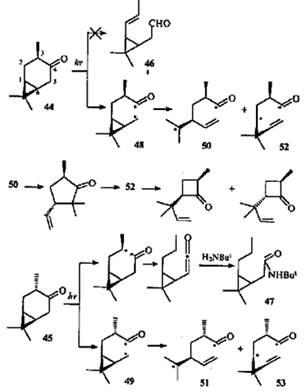
Вопреки ожиданиям, не наблюдалось образования альдегида 46
и эпимеризации кетона 45
. Однако при облучении последнего в присутствии трет
-бутиламина в продуктах реакции было найдено небольшое (>1%) количество амида 47
. Это свидетельствует о том, что гомолитический разрыв связи С(3)—С(4) все-таки происходит, но в крайне незначительной степени.
Формально фотохимическое поведение кетонов 44
и 45
очень похоже на аллильные перегруппировки β,γ-ненасыщенных кетонов.45
Кропп с соавторами считал фотохимическую перегруппировку каранонов 44
и 45
первым примером циклопропильного аналога фотохимической аллильной перегруппировки.44
Однако в случае β,γ -ненасыщенных кетонов реакция обратима, а в случае кетонов 44
и 45
— нет.
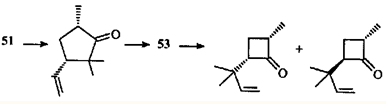
По аналогии с β,γ -ненасыщенными кетонами можно полагать, что направление α-расщепления углеродных связей обусловлено "аллильным" влиянием ЦПК, ослабляющим связь С(4)—С(5) за счет эффекта сопряжения или индуктивного. В образовавшихся радикалах 48
и 49
должны раскрыться связи С(1)—С(6) или С(1)—С(7). Соотношение присутствующих в продуктах реакций циклопентанонов и циклобутанонов (3:1) отражает преимущественное раскрытие более замещенной связи С(6)—С(7) и относительную стабильность третичного (50, 51
) и вторичного (52, 53
) радикалов.
Есть и еще одно отличие аллильных фотохимических перегруппировок β,γ-ненасыщенных кетонов от аналогичных циклопропильных. Аллильная перегруппировка считается согласованным процессом.45
Но превращение каранонов 44
и 45
, по мнению авторов,44
не может быть согласованной реакцией, по крайней мере об этом свидетельствует образование эпимерных циклобутанонов.
Направление скелетной фотохимической перегруппировки терпеноидов ряда карана зависит от ориентации заместителей: прямое облучение транс
-3-метокси-4-каранона (54
) в этаноле протекает с раскрытием шестичленного цикла по связи С(3)—С(4) (что для β,γ-циклопропилкетонов бывает крайне редко) и присоединением этанола.39
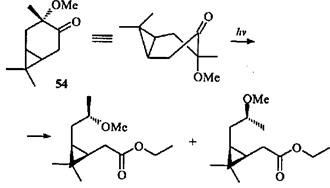
В случае цис
-3-метокси-4-каранона (55
) с экваториальной метоксигруппой наблюдается исключительно элиминирование II типа по Норришу и образование каранона 44
; требуемое плоское шестичленное переходное состояние (ПС) А
легко достижимо, и образование кетона 44
успешно конкурирует с расщеплением связи С(3)—С(4).39
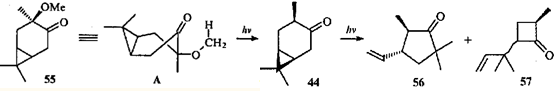
Кетон 44
под действием света подвергается гомолитическому разрыву связи С(4)—С(5) и гомоаллильной перегруппировке, характерной для β,γ -циклопропилкетопов.44,46
При этом образуются циклопентанон 56
и циклобутанон 57
.
Таким образом, перегруппировки карановых монотерпеноидов под действием света можно разделить на две группы: перегруппировки, в которых в качестве хромофора выступают элементы углеродного скелета — циклопропан и двойная связь, — и перегруппировки, в которых хромофором является функциональный заместитель. Применение сенсибилизаторов приводит к перегруппировкам первого типа с образованием производных бицикло[3.2.0]гептана. Воздействие облучения в отсутствие сенсибилизатора, как правило, приводит к перегруппировкам второго типа, при этом возможны разрыв большого или малого циклов, сужение большого цикла или образование трициклической структуры. Направление реакции в каждом конкретном случае определяется природой и положением заместителя, являющегося хромофором.
IV. Согласованные реакции
Если ионным реакциям соединений каранового ряда посвящено множество работ, которые неоднократно систематизировались, то согласованные превращения значительно менее исследованы, и обобщение этого материала до сих пор не делалось. Справедливости ради следует отметить, что далеко не все реакции, которые формально относятся к согласованным, оказываются таковыми в действительности. Например, в случае 2,4-карадиена (58
) 1,5-термический сдвиг формально является согласованной сигматропнои реакцией. Однако, во-первых, она протекает не через циклическое ПС (с одновременным разрывом и образованием связей С—С), а через дирадикальный интермедиат, а во-вторых, по сути, она не является сигматропным процессом. Иногда согласованный механизм осуществляется через сильно поляризованное ПС или сопряжен с ионным механизмом. Обзор подобных реакций карановых монотерпеноидов особенно интересен потому, что именно в них иногда осуществляются уникальные превращения, характерные только для этих соединений (например, перегруппировка Берсона-Вилькотта 47
). Исследование согласованных реакций существенно расширяет наши представления о механизмах химических реакций вообще.
1. Интерконверсия норкарадиен - циклогептатриен
Одно из интереснейших скелетных превращений карановых структур — перегруппировки, связывающие терпеноиды ряда карана и ряда 1,1,4-триметилциклогептана, — привлекло внимание исследователей и привело к открытию такого замечательного явления в органической химии, как "быстрое" равновесие норкарадиен - циклогептатриен. Этой теме посвящено большое количество работ последней трети двадцатого века.47-55
Быстрое равновесие валентных таутомеров норкара-2,4-диена (59
) и циклогепта-1,3,5-триена (60
) обусловлено типично согласованной реакцией — дисротаторным раскрытием и замыканием трехчленного карбоцикла, причем этот процесс подчиняется правилам сохранения орбитальной симметрии, сформулированным Вудвордом и Гофманом.56
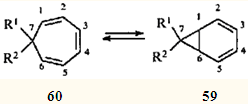
Для незамещенного углеводорода (R1
= R2
= H) это равновесие настолько сдвинуто в сторону моноциклического таутомера 60
, что норкарадиен не удавалось обнаружить никакими физическими методами. Его существование постулировали на основании поведения замещенных аналогов (R1
= CN, R2
= CF3
; R1
= CO2
Me, R2
== Ph; R1
= CO2
Me, R2
= C6
H4
OMe-4), которые в виде равновесных смесей с соответствующими циклогептатриенами обнаружены при исследовании последних методом низкотемпературного ЯМР.55
Только в 1981 г., четверть века спустя после появления гипотезы о валентной таутомерии циклогептатриена, удалось зафиксировать незамещенный норкарадиен при низкотемпературном (77 К.) фотолизе трицикло[3.2.2.02,4
]нон-6-енди-8,9-она.57
Экспериментально установлено, что бициклическую структуру способны стабилизировать π-акцепторные заместители в положении 7.55
Природу такой стабилизации изящно объяснил Р.Гофман в рамках метода граничных МО.53
Очевидно, что метальные заместители стабилизаторами быть не могут, и поэтому для системы 2,4-карадиен (58
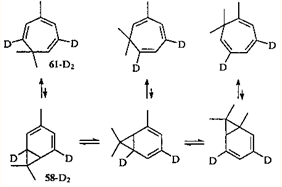
) — 3,7,7-триметилциклогепта-1,3,5-триен (61
) равновесие всегда сдвинуто в сторону моноциклического партнера. Однако эти валентные таутомеры — все-таки разные, реально существующие соединения; они обладают различной реакционной способностью, что должно проявляться (и проявляется, как будет показано ниже) в химических реакциях.
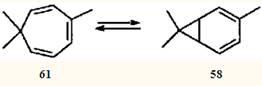
Перегруппировки, обусловленные описанной выше валентной таутомерией, будут обсуждены более подробно, чем ионные, поскольку в отечественной научной литературе проблема таутомерии норкарадиена практически не рассматривалась. К сожалению, даже исследователи, работающие в области химии терпеноидов, привлекают для объяснения расширения каранового скелета до циклогептанового недостаточно обоснованные гипотезы.12,58
Перегруппировка циклогептатриен — норкарадиен была обнаружена при изучении химических превращений эйкарвона (2,6,6-триметилциклогепта-2,4-диенона (62
)) — терпеноида, впервые синтезированного Байером,59
но впоследствии обнаруженного в эфирных маслах различных растений.60
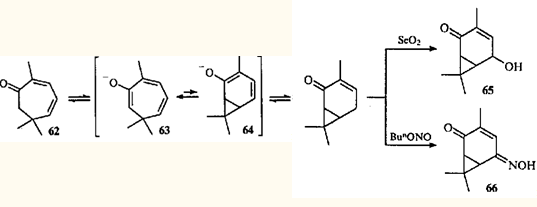
Гипотеза о валентной таутомерии моно- и бициклических енолов 63
и 64
, как возможное объяснение скелетной перегруппировки кетона 62
при образовании 3,7,7-триметил-5-гидрокси-3-карен-2-она (65
) и 3,7,7-триметил-5-оксиимино-3-карен-2-она (66
), была выдвинута в 1956 г.61,62
Впоследствии оказалось, что гораздо более распространенными являются перегруппировки карановых производных в триметилциклогептановые, поскольку равновесие в динамическом взаимопревращении двух таутомеров сильно сдвинуто в сторону моноциклического партнера.63
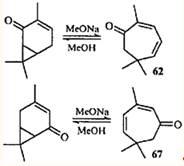
2,5,5-Триметилциклогепта-2,4-диенон (67
), так же как и кетон 62
. обнаружен в скипидарах из Pinus silvestris L
.64
По-видимому, присутствие этих кетонов в живичном скипидаре объясняется тем, что из 3-карена (одного из основных компонентов скипидара) в процессе развития растения образуются непредельные кетоны ряда карана, которые затем согласованной электроциклической реакцией превращаются в моноциклические диеноны. Подтверждением этого могут служить результаты жидкофазного окисления 3-карена: содержание кетонов 62
и 67
в продуктах реакции достигало 20 и 28%.65
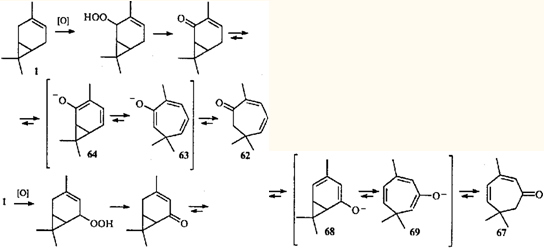
Следует отметить, что перегруппировки 64 →
63
и 68 →
69
— единственное на сегодняшний день объяснение присутствия в эфирных маслах кетонов с семичленным циклом. Общая схема биогенезиса терпенов 66,67
вообще не предусматривает образования подобных соединений. Однако широкое распространение в природе 3-карена и его производных и тот факт, что триметилциклогептановые соединения присутствуют только в тех эфирных маслах, где есть терпепоиды ряда карана, можно считать косвенным доказательством предположения о путях образования терпеноидов с семичленным циклом. Гораздо менее правдоподобным, по нашему мнению, является предположение о возможности образования эйкарвона (62
) в процессе биосинтеза 12
тем же путем, каким его получал Байер: гидробромированием карвона и последующим дегидробромированием образовавшегося 8-бром-1-ментен-6-она.59
Наконец, серьезным аргументом в пользу перегруппировки 58 → 61
как причины образования циклогептановых терпеноидов не только in
vitro
, но и in
vivo
, служит обнаружение триена 61
в сосновом живичном скипидаре и в живицах отдельных деревьев Pinus
silvestris
.68
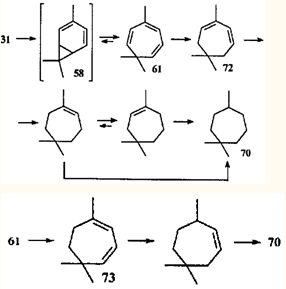
Поскольку этот триен существует только в паре с кара-2,4-диеном, который присутствует в скипидаре и живице, можно сделать вывод о возможности каталитического (ферментативного) дегидрирования 2-карена, также присутствующего в скипидарах.
Двойственная природа триена 61
чрезвычайно наглядно проявляется в реакции гидрирования (H2
, Pt, 20°С); при неполном гидрировании в продуктах реакции наряду с триметилциклогептадиенами и триметилциклогептенами обнаруживаются также 3-карен (1
) и 2-карен (31
). Продукт исчерпывающего гидрирования представляет собой смесь 1,1,4-триметилциклогептана 70
(96%) и карана 71
(4%).69

Разумеется, соотношение конечных продуктов гидрирования (70:71
= 96:4) не отражает содержания таутомеров в исходной равновесной смеси. Сравнительно большое количество карана 71
может быть обусловлено тем, что плоская диеновая система карадиена 58
успешно конкурирует с неплоскими диеновыми фрагментами триена 61
в реакции 1,4-присоединения водорода.
Необычным является гидрирование 3-карена. В 1966 г. Кокером с соавт.70
было установлено, что продукт гидрирования 3-карена на платине представляет собой смесь соединений 71
(98%) и 70
(2%), а при гидрировании на палладии преобладающим продуктом является углеводород 70
(74%).70
Позже оказалось, что гидрирование 3-карена на медно-никелевом катализаторе протекает с образованием исключительно 1,1,4-триметилциклогептана (70
).58
Авторы обеих работ считали, что такое течение реакции обусловлено изомеризацией 3-карена (1
) в 2-карен (31
) и 1,4-присоединением водорода к винилциклопропановой системе последнего с образованием углеводорода с семичленным циклом.
При этом подразумевалось, что возможность или невозможность 1,4-присоединения, которое является ключевой стадией перегруппировки, зависит от природы катализатора. Но 1,4-присоединение возможно только в том случае, если имеется сопряжение в диеновом (или, для углеводорода 31
, в гомодиеновом) фрагменте. Однако молекула терпена 31
находится в конформации "плоское кресло",38
что ставит под сомнение возможность такого сопряжения. Фотоэлектронный спектр показывает, что π-орбиталь и ВЗМО диметилциклопропанового кольца в молекуле соединения 31
практически не взаимодействуют.17
То что 1,4-присоединение для молекулы 2-карена (31
) неосуществимо, подтверждено экспериментально: взаимодействие синглетного кислорода — исключительно активного диенофила — с этим терпеном приводит лишь к аллильным гидропероксидам,27
в то время как его реакции, например, с α-фелландреном и α-терпиненом,71
дают продукты 1,4-присоединения — эндопероксиды.
При обсуждении механизма реакции Кокер с соавт.70
не учитывал полученные им же результаты: при использовании палладиевого катализатора содержание углеводорода 70
увеличивалось с повышением температуры гидрирования и составляло 100% при температурах выше 70°С, т.е. на соотношение продуктов реакции прежде всего влияет температура. Аналогичная температурная зависимость позже была установлена и для гидрирования на платине: если при 20°С содержание углеводорода 70
составляет 2-3%, то при 70 и 90°С — 37 и 46% соответственно.72
Скелетная перегруппировка 3-карена при гидрировании становится объяснимой, если учесть, что и платина, и палладий катализируют как прямую, так и обратную реакции и способны осуществлять дегидрирование в присутствии свободного водорода.73
Гидрирование 3-карена в результате присоединения водорода к двойной связи приводит к карану; дегидрирование 2-карена, всегда имеющегося в реакционной среде (вследствие изомеризации 3-карена, которая происходит на катализаторе в присутствии водорода), обусловливает появление диена 58
, триена 61
и других соединений с измененным скелетом.74
Повышение температуры благоприятствует эндотермической реакции дегидрирования и, соответственно, увеличению содержания перегруппированных продуктов. Следует отметить, что гидрирование на медно-никелевом катализаторе, когда углеводород 71
был единственным продуктом, также проводили при высокой температуре (180°С).58
Прямым доказательством реализации именно этого механизма служит диспропорционирование 2- и 3-каренов на палладии и платине в отсутствие свободного водорода. В этом случае среди продуктов был идентифицирован триен 71
, что подтверждает предположение о дегидрировании 2-карена (31
) как о стадии, отвечающей за образование перегруппированных продуктов.75
В реакционной смеси присутствовали также триметилциклогептадиены 72
и 73
. Однако ни один из известных путей изомеризации терпенов 1
и 31
(в том числе и каталитический) не приводит к подобному расширению цикла.
На основании сказанного выше можно сделать вывод, что перегруппировка карановых структур в триметилциклогептановые происходит в том случае, когда в ходе реакции возможно образование диена 58
(или его производных). Проще всего получить карадиен 58
из производных 2- и 4-карена, имеющих функциональную группу в аллильном положении. Так, при дегидрохлорировании 4-хлор-2-карена (74
) образуется триен 61
.76
В продуктах превращения эпоксида (75
) в присутствии рениевых катализаторов обнаружили 4-карен-3-ол (76
), который при дегидратации также дает углеводород 61
.77
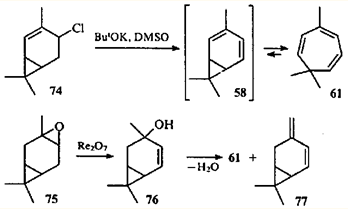
Теоретически дегидратация спирта 76
может идти и по другому направлению: с образованием терминальной метиленовой группы. Соответствующий углеводород — 3(10),4-карадиен (77
) — действительно был обнаружен в продуктах реакции эпоксида 75
.77
Углеводород 77
, полученный впервые дегидрохлорированием 4-хлор-3(10)-карена (78
),78
является первым устойчивым диеном с карановым скелетом.
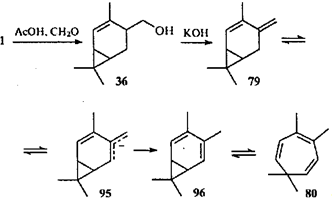
Дегидратацией спирта 36
в присутствии гидроксида калия был синтезирован второй представитель этого ряда — 4-метил-3(10),4-карадиен (79
).79
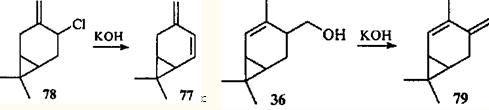
Диены 78
и 79
устойчивы (т.е. не склонны к таутомерным превращениям), так как в отличие от соединения 58
они имеют трансоидную диеновую систему, для которой невозможно электроциклическое замыкание и, следовательно, согласованная перегруппировка. Однако эти соединения весьма реакционноспособны: даже слабые кислоты, такие как салициловая и бензойная, изомеризуют трансоидную диеновую систему в цисоидную; затем происходит согласованная перегруппировка в циклогептатриены.80
При дегидратации спирта 36
над Al2
O3
(кислотой Льюиса) в продукте реакции не обнаруживается даже следов диена 79
(в отличие от дегидратации над щелочью79
), но присутствует 3,4,7,7-тетраметилциклогепта-1,3,5-триен (80
).81
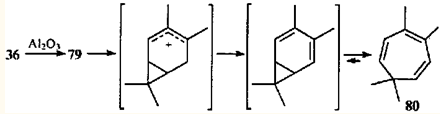
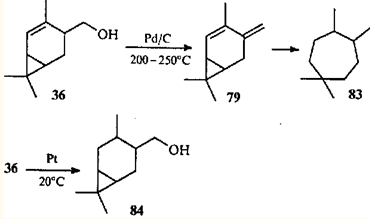
Гидрирование диена 79
на платиновой черни при 20°С также протекает с перегруппировкой, что, очевидно, обусловлено его изомеризацией на катализаторе.80
В данном случае примечательно сравнительно высокое содержание в продукте исчерпывающего гидрирования соединений с бицикло[4.1.0]гептановым скелетом 81
, 82
. Это означает, что скорость присоединения водорода к диену 79
выше, чем скорость его изомеризации, которая является побочным процессом (по крайней мере, при комнатной температуре).

Сочетание трех реакций — дегидратации, изомеризации и гидрирования — приводит к тому, что продуктом каталитического гидрирования спирта 36
в жестких условиях является 1,1,4,5-тетраметилциклогептан (83
).82
На платине при 20°С реакция протекает без изомеризации как простое присоединение водорода к двойной связи и дает 4-гидроксиметилкаран (84
).83
Перегруппировка, обусловленная валентной таутомерией, позволяет получать функциональные производные циклогептатриена. Так, диен 79
в реакции Принса дает два продукта гидроксиметилирования — моно- (85
) и бициклический (86
).84
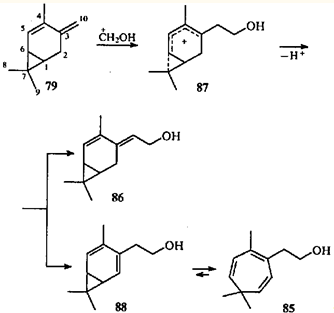
При электрофильной атаке терминальной двойной связи в соединении 79
протонированной молекулой формальдегида образуется гомодиенильный ион 87
, который стабилизируется двумя путями: выбросом протона от атома С(5) с образованием спирта 88
или от атома С(10) с образованием аллильного спирта 86
. Спирт 86
под действием кислоты изомеризуется в карадиеновый спирт 88
, который затем перегруппировывается в спирт 85
.
Моноциклический спирт 85
может образоваться и по другому механизму. Если к исходному углеводороду присоединяется молекула формальдегида, первая стадия реакции протекает как согласованная через шестичленное поляризованное ПС (89
)85
и приводит к бициклическому спирту 88
. Последующая перегруппировка дает продукт реакции 85
. Таким образом, превращение диена 79
в триеновый спирт 85
может протекать как последовательность двух согласованных реакций.
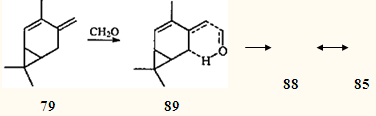
Следует отметить, что атака электрофильного агента (и заряженной молекулы, и диполя) направлена на атом С(10) соединения 79
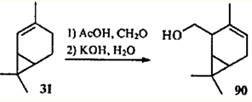
, т.е. на один из концов сопряженной гомотриенильной системы, более доступный с пространственной точки зрения. Однако в реакции с 2-кареном (31
) протонированная молекула формальдегида присоединяется к наименее замещенному концу двойной связи, давая гомоаллильный спирт 90
,86
что является дополнительным подтверждением отсутствия сопряжения в молекуле 2-карена.
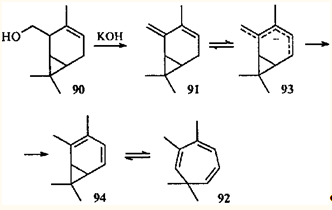
Следует отметить, что основным продуктом дегидратации 2-гидроксиметил-3-карена (90
) гидроксидом калия является не карадиен с экзоциклической двойной связью (91
), а 2,3,7,7-тетраметилциклогепта-1,3,5-триен (92
), который, очевидно, образовался через интермедиаты 93, 94
.86
При дегидратации спирта 36
среди продуктов реакции кроме триеиа 80
обнаружен диен 79
.84
В пентадиенильном анионе 93
заряд делокализован в большей степени, чем в аллильном анионе 95
, поэтому превращение диена 91
в конечный продукт 92
происходит легче, чем превращение диена 79
в диен 96
и далее в циклогептатриен 80
.
Вообще говоря, согласованная перегруппировка трансоидных карадиенов происходит чрезвычайно легко, поэтому, чтобы ее избежать и получить весьма лабильный неперегруппированный диен, применяют специальные приемы. Так, углеводород 91
удалось синтезировать из спирта 90
только путем термического разложения N
-оксида 97
.86
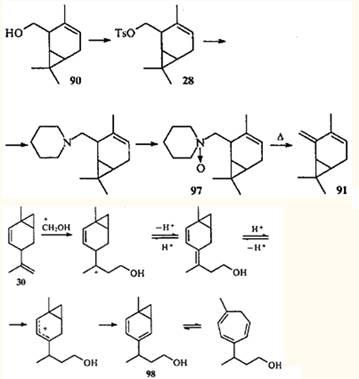
Возвращаясь к реакции Принса, можно отметить, что в тех случаях, когда молекула углеводорода, имеющая бицикло[4.1.0]гепт-2-еновую систему, содержит дополнительную изолированную двойную связь (как в соединении 30
), первоначальная атака формальдегида направляется на эту связь. Однако в условиях кислотного катализа первоначально образующийся бициклический спирт легко изомеризуется, давая бицикло[4.1.0]гепт-2,4-диеновую систему (например, 98
), в которой происходит согласованная перегруппировка.86
2. Сигматропные перегруппировки
Термическая перегруппировка 2-карена (31
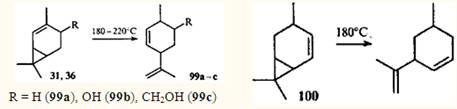
) и его производных была первым примером превращений, которые впоследствии стали называть 1,5-гомодиенильным сдвигом.87
Хорошо известно, что при температурах выше 330°С винилциклопропан превращается в циклопентен; реакция протекает путем гомолитического разрыва связи, изомеризации образовавшегося дирадикала и последующей рекомбинации.88
Энергия активации этой перегруппировки довольно высока и составляет 208 кДж•моль-1
(см.89
).
Однако 2-карен и его производные, молекулы которых содержат винилциклопропановый фрагмент, претерпевают перегруппировку уже при 180°С, образуя при этом соответствующие производные транс
-изолимонена 99а-с.
90-91
Подобным же образом реагирует 4-карен (100
).92

Это превращение обусловлено особенностями строения производных 2- и 4-карена: подобно тому, как в цис
-1,3-диеновых системах возможен 1,5-диенильный сдвиг водорода, в соединениях ряда карана, имеющих винилциклопропановый фрагмент, возможен 1,5-гомодиенильный сдвиг. Реакция протекает через шестиэлектронное переходное состояние и подчиняется правилам отбора для термических водородных сдвигов.56
Гомолитического разрыва углерод-углеродной связи не происходит, так как энергия активации 1,5-гомодиенильного сдвига значительно ниже, чем энергия винилциклопропановой перегруппировки; в случае 2-карена она составляет 118 кДж•моль-1
(см.91
). Необходимое условие осуществления 1,5-сдвига — эндо
-ориентация участвующих в образовании ПС связи С—Н и винильной группы.93
Это — единственное стереохимическое требование для термической перегруппировки цис
-1-метил-2-винилциклопропана, расположение заместителей в молекуле которого не вносит дополнительного напряжения. Это требование выполняется и для молекулы 2-карена (31
), но из-за того, что двойная связь входит в шестичленный цикл, она отклоняется от направления, оптимального для сопряжения с ЦПК и образования ПС. Расстояние между С(3) и атомом водорода при С(8) в конформации "плоского кресла" составляет ~3.1 Å.38
Это означает, что молекула 2-карена (31
) при нагревании должна сначала изменить конформацию (при этом расстояние С(3)—Н(8) уменьшается до 2.З Å), и только затем может произойти 1,5-гомодиенильный сдвиг.
Природа заместителя в положении 4 может влиять на энергию активации конформационного перехода, но даже объемные заместители не препятствуют 1,5-сдвигу; правда термическая перегруппировка таких соединений требует более высокой температуры.94
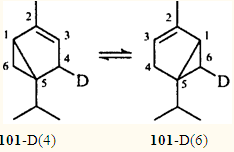
α-Туйен (101
), молекула которого также содержит винилциклопропановый фрагмент, при нагревании выше 300°С остается неизменным. Однако эксперименты с дейтериевой меткой показали, что в действительности происходит вырожденная винилциклопропановая перегруппировка. В данном случае ПС, необходимое для осуществления 1,5-гомодиенильного сдвига, является труднодостижимым: расстояние между D(4) и С(5) или С(3) составляет ~ 2.8 Å.95
Следует отметить, что благодаря геометрии молекул именно карановые структуры оказались подходящими моделями для изучения 1,5-гомодиенильного сдвига. В реакциях термической изомеризации терпеноидов с бицикло[4.1.0]-гепт-2-еновым скелетом 1,5-гомодиенильный сдвиг происходит регио- и стереоспецифично, что позволяет, например, синтезировать новые кислородсодержащие терпеноиды из производных 2-карена.96
Не всегда наличие эндо
-ориентированного винилциклопропанового фрагмента и подходящей связи С—Н в соединениях каранового ряда означает возможность термического 1,5-водородного сдвига. Так, при нагревании карадиена 77
в бензоле в интервале температур 120-190°С основным (при сравнительно низких температурах — единственным) продуктом его превращения был триен 61
.97
При повышении температуры до 170 - 190°С в небольших количествах (до 3 %) образовывались также п
- и м
-цимолы.
Казалось бы, наиболее реальным процессом в описываемых условиях должен быть 1,5-гомодиенильный сдвиг: винильная группа и одна из связей С(8)—Н имеют эндо
- ориентацию, расстояние С(4)—Н(8) составляет ~2.2Å, т.е. легко достижимо идеальное ПС 1,5-сдвига, рассчитанное теоретически.93
Однако продуктом такого превращения должен быть м
-мента-1(7),4,8-триен, который в действительности не образуется. Винилциклопропановая перегруппировка при этих температурах также невозможна, так как для радикальной реакции они недостаточно высоки. Можно предположить, что за скелетную перегруппировку ответственна валентная таутомерия. Тогда термическая изомеризация 77
→ 61
фактически сводится к превращению трансоидного диена 77
в цисоидный диен 58
. Формально — это 1,3-сигматропный сдвиг.

Принято считать, что некатализируемый 1,3-сдвиг водорода в термических реакциях не происходит, так как переходное состояние разрешенного антараповерхностного сдвига является труднодостижимым из-за больших угловых напряжений в пропиленовом фрагменте.98
Супраповерхностный сдвиг, который вполне мог бы реализоваться с пространственной точки зрения, запрещен правилами сохранения орбитальной симметрии. Однако расчет ab
initio
1,3-сдвига в пропене показал, что запрещенный супраповерхностный перенос водорода требует меньшей энергии активации, чем разрешенный антараповерхностпый.99
Для объяснения этого неожиданного результата была привлечена гипотеза о контроле орбиталью, лежащей ниже граничной.100
Эта гипотеза лежит в основе предположения об осуществлении 1,3-сдвига в молекуле диена 77
.
К исследованию механизма термической изомеризации карадиена 77
были привлечены кинетические исследования.97
Превращения углеводорода 77
в триен 61
— последовательность двух согласованных реакций, причем первая (предполагаемый 1,3-сдвиг) по совершенно очевидным соображениям является лимитирующей. Кинетические исследования показали, что эта стадия описывается уравнением первого порядка. Определенная из экспериментальных данных энергия активации составляет 112 кДж•моль-1
, что близко к расчетной величине энергии активации антараповерхностного 1,3-сдвига (115 кДж•моль-1
).99
На то, что 1,3-сдвиг возможен в этой реакции, указывает высокое отрицательное значение энтропии активации, равное 96 кДж•моль-1
•град-1
(—23 э.е.). Сравнение этой величины с соответствующим параметром 1,5-сдвига (-8 ÷ -16 э.е.) показывает, что переходное состояние термической перегруппировки карадиена 77
является высокоупорядоченным и участвующие атомы связаны очень жестко. Энергия активации 1,5-сдвига в 2-карене (31
) (118 кДж•моль-1
)91
несколько выше, чем энергия активации термической изомеризации диена 78
, и это может быть объяснением, почему в данном случае не осуществляется 1,5-сдвиг.
Особенно интересна термическая перегруппировка Берсона - Вилькотта.47
Формально она представляет собой перемещение метальной группы по всем возможным положениям триеновой системы в молекуле углеводорода 61
. Тем не менее её следует рассматривать в ряду карановых перегруппировок, Так как в действительности осуществляется не 1,3- или 1,5-алкильпый сдвиг, а 1,5-сдвиг диметилметиленовой группы в карадиеновом таутомере 58
, что было показано с помощью дейтериевых меток.
Эта частная реакция послужила толчком для развития экспериментальных и теоретических исследований, которые значительно расширили и в чем-то изменили классические представления о сигматропных реакциях. Триен 61
оказался прекрасной моделью для этих исследований, так как перегруппировка осуществляется только при наличии гем
-диметильной группы: в отсутствие геминальных заместителей циклогептатриен претерпевает 1,5-сдвиг водорода уже при температурах выше 100°С.101
Присутствие третьей метильной группы, которая "гуляет" по триеновой системе семичленного цикла, позволило обнаружить эту перегруппировку ("walkrearragement").
Следует отметить, что механизм сдвига не позволяет считать перегруппировку гептатриена 61
в полном смысле согласованной, ибо она протекает через дирадикальный интермедиат при температурах ~ 400°С. В процессе перегруппировки происходит перемещение циклопропановой связи, которая в исходной молекуле не является σ-связью, что тоже не позволяет считать перегруппировку сигматропной. Тем не менее довольно долго полагали, что перегруппировка триена 61
полностью отвечает правилам отбора для согласованных сигматроппых миграций алкильных групп, т.е. протекает как супраповерхностная с сохранном конфигурации мигрирующей группы. Изящные работы Клернера с соавт.102-104
полностью опровергли эти представления: заменив одну из метильных групп алкоксикарбонильной, нитрильной или алкоксиалкильной, он доказал, что перегруппировка протекает с обращением конфигурации, т.е. по "запрещенному" пути.
Недавно в вышедших почти одновременно теоретических работах 105,106
это явление было объяснено на основании расчетов ab
initio
.
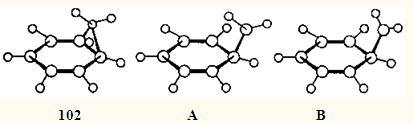
В случае незамещенного норкарадиена 102
в дирадикальном ПС А
запрещенного термического 1.5-алкильного сдвига метиленовая группа с неспаренным электроном находится несколько дальше от шестичленного кольца, чем в ПС В
разрешенного сдвига, что понижает энергию ПС запрещенного сдвига на ~4 кДж•моль-1
(см.105
).
Переходные состояния C
и D
для 1,5-сдвига в цис
- (103
) и транс
-7-циано-7-метилноркарадиенах (104
), протекающего с обращением конфигурации (и поэтому запрещенного), на 12 и 13 кДж•моль-1
соответственно более предпочтительны, чем ПС Е
аналогичного сдвига с сохранением конфигурации (и поэтому разрешенного). Расстояние между атомами водорода метильной группы и цикла в запрещенных ПС С
и D
на 0.12 и 0.37Å меньше, чем в разрешенном ПС Е
.106
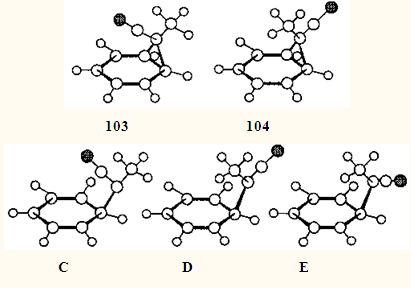
Можно сделать вывод, что при протекании сигматропной перегруппировки через дирадикальный интермедиат стерический фактор может доминировать над электронным, что и приводит к нарушению фундаментальных правил сохранения орбитальной симметрии. Исследования, приведшие к этому поразительному результату, были начаты с изучения терпеноидов — триеиа 61
и его валентного таутомера диена 58
.
3. Термическое элиминирование
При попытке дегидратировать спирт 36
с помощью борной кислоты вместо ожидаемого диена 79
была получена смесь углеводородов 30,105
и 106
.107
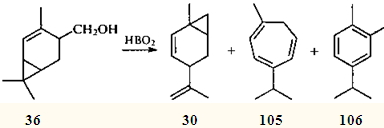
Позже было установлено, что эти углеводороды образуются также при термическом разложении моно-, ди- и тризамещенных эфиров борной кислоты и спирта 36
(промежуточных продуктов описанной выше дегидратации), причем первоначально образуется бициклический углеводород 30
; были идентифицированы и другие продукты его превращения.108
Подобным же образом реагировали тозилат и хлорацетат спирта 36
; ацетат и аминоацетат претерпевали 1,5-гомодиенильный сдвиг.109
Термическое разложение сложных эфиров — хорошо изученная реакция, которая является одним из способов получения олефинов за счет отщепления кислотного остатка и атома водорода, связанного с β-углеродным атомом. Элиминирование протекает через циклическое шестиэлектронное (ароматическое) ПС. Однако в случае спирта 36
и его производных пиролиз происходит не с образованием двойной связи, а с разрывом одной из связей гем
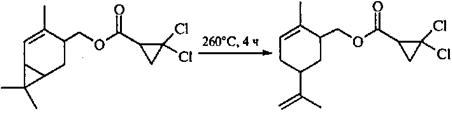
-диметилциклопропанового кольца с одновременным замыканием нового трехчленного карбоцикла. С целью изучения механизма такой необычной перегруппировки были проведены дополнительные исследования: изучена кинетика термического разложения тозилата 25
в пиридине37
и его сольволиз.35
Эти исследования позволили предположить механизм перегруппировки.37
Она протекает как согласованная, через ароматическое переходное состояние, но, в отличие от классического шестиэлектронного, это ПС является десятиэлектронным. Хотя гипотеза о десятиэлектронном ПС ранее в органической химии не использовалась, она не имеет принципиальных внутренних противоречий. Недавно методом B3LYP/6-31G* было рассчитано десятичленное ПС для [5,5]-сигматропной перегруппировки Коупа.110
Расчет методом функционала плотности показывает, что десятичленное ПС может быть неплоским, сохраняя при этом свою ароматичность.111
Причиной такого необычного пути реакции является пространственный фактор. Для образования шестичленного ПС при элиминировании элементов кислоты кислотный остаток в эфире 107
должен иметь заслоненную конформацию по отношению к атому водорода Н(4). Это создает возможность сближения атома кислорода кислотного остатка и одного из атомов водорода эндо
-метильной группы, связанной с циклопропановым кольцом, и обеспечивает участие винилциклопропанового фрагмента в образовании переходного состояния. Для выполнения условия ароматичности (соблюдения правила Хюккеля) требуется перекрывание p
-орбиталей атомов, не являющихся соседними (С(3), С(11)). Образование циклопропанового кольца и одновременное преобразование винилциклопропанового фрагмента в 1,4-диеновую систему происходит в результате такого перекрывания через пространство.

Эта точка зрения получила экспериментальное подтверждение.37
Детальное исследование разложения эфиров 107-109
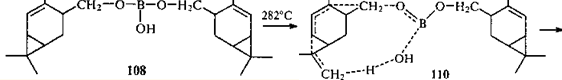
показало, что в этих реакциях образуются продукты разного состава. Так, среди продуктов пиролиза эфира 108
присутствовала также вода (3-5%).
→ 30
+ 107
+ H2
O
Первая стадия пиролиза протекает как фрагментация молекулы диэфира 108
через десятичленное ПС 110
. включающее гидроксильную группу. При этом образуются бициклический углеводород 30
, вода и эфир 107
. Далее эфир 107
разлагается по описанной выше схеме с образованием борной кислоты и второй молекулы углеводорода 30
. Среди продуктов пиролиза эфира 109
, помимо углеводорода 30
и продуктов его превращения, присутствовали 2- и 3-карены, ментадиены, а также спирт 36
:

На первой стадии кроме углеводорода 30
образуется эфир 107
, который разлагается по описанной схеме, и спирт 36
. Последний в условиях реакции (380°С) частично деформилируется. давая 3-карен (1
).91
Кислотная изомеризация этого терпена приводит к появлению среди продуктов 2-карена и ментадиенов.
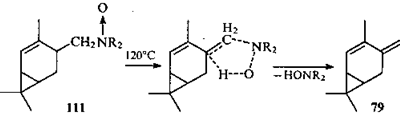
Следует отметить, что при пиролизе N
-оксида 111
, который близок по строению эфирам 107-109
, с количественным выходом образовался углеводород 79
, содержащий экзоциклическую двойную связь.112
Этот результат можно рассматривать как косвенное доказательство решающей роли пространственного фактора в таких перегруппировках.
В оксиде 111
функциональная группа имеет на одно звено меньше, чем в эфирах 107-109
, за счет чего появляется возможность образования пятичленного (но шестиэлектронного, т.е. по-прежнему ароматического) ПС, меньшего по размерам, в котором атом С(4) имеет скошенную конформацию.
Несомненно ПС термического разложения эфиров сильно поляризовано; структура ПС зависит от природы образующих его атомов. Поэтому ацетат, борат, тозилат спирта 36 проявляют в этой реакции различную реакционную способность. На реакционной способности сказывается даже вторичное влияние гетероатомов: ацетат и аминоацетат претерпевают 1,5-гомодиенильный сдвиг, термолиз тозилата происходит при 90°С, хлорацетата — при 180°С. моно-, ди- и трехзамещенных боратов — при 225, 282 и 380°С соответственно.113
Борный эфир 112
, в котором заведомо невозможно 1,2-элиминирование, пиролизуется с образованием 3-карена (1
). Предполагают, что последний образуется в результате кислотной изомеризации первоначального продукта пиролиза — трициклического углеводорода 113
. Ни один из опробованных в этой реакции эфиров 3,6,6-триметилбицикло[3.1.0]гекс-3-илметанола (тозилат, ацетат, амино- и хлорацетаты) не вел себя подобным образом.

В разделе II
.2 мы отмечали, что сольволиз тозилатов 25
и 28
приводит к одному и тому же продукту 29
.36
При пиролизе этих соединений также образуется один и тот же продукт реакции. 30
, правда пиролиз эфира 28
происходит при более высокой температуре (180-200°С), чем эфира 25
(70 - 100°С). В качестве возможного объяснения такого результата мы предполагаем предварительный 1,3-сдвиг тозилоксиметильной группы, который происходит при высокой температуре. Подобный сдвиг ацильной группы наблюдался при фотолизе кетона 42
43
(см. раздел III
).
4. Ионные реакции и валентная таутомерия
Выше уже упоминались ионные превращения карановых монотерпеноидов (например, соединений 74
и 76
), приводящие к образованию 2,4-карадиеновой структуры, которая в результате электроциклического раскрытия трехчленного цикла перегруппировывается в триметилциклогептатриеновую. Скелетная перегруппировка в этих случаях происходит за счет валентной таутомерии. Для соединений ряда карана, имеющих более сложный углеродный скелет, возможны ионные пере группировки другого типа в результате которых образуются структуры, склонные к таутомерным превращениям.
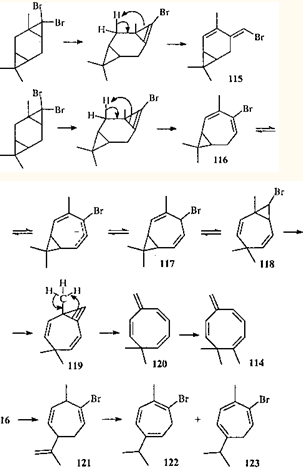
Так. аддукт 3-карена с дибромкарбеном при действии суперосновной системы KOR-DMSO (R=Н, But
) селективно дегидробромируется с образованием 1,8,8-триметил-5-метиленциклоокта-1,3,6-триена (114
).114
При уменьшении конценграции дегидробромирующего агента из реакционной смеси были выделены и идентифицированы и другие продукты, что позволило предложить схему перегруппировки.115
Отщепление первой молекулы бромистого водорода сопровождается изомеризацией трехчленного цикла с образованием бромидов 115
и 116
. Бромид 115
с сохраненным карановым скелетом образуется в результате раскрытия трехчленного цикла по внешней связи и не подвергается дальнейшим превращениям под действием системы KOH-DMSO. Соединение 116
изомеризуется, давая бромид 117
. в котором имеется цис
-дивинилциклопропановая система. В нем происходит перегруппировка Коупа116
путем согласованного раскрытия ЦПК. приводя к валентному таутомеру 118
. Дегидробромирование соединения 118
ведет к циклопропену 119
, который изомеризуется в более устойчивое циклооктатриеновое производное 120
. Последнее в условиях реакции метилируется димсил-анионом.
В ходе этой реакции происходит еще одна согласованная перегруппировка. Бромид 121
образуется в результате 1,5-гомодиенильного сдвига; дальнейшая ионная изомеризация этого соединения приводит к бромидам 122
и 123
.
5. Сигматропные сдвиги в заряженных системах
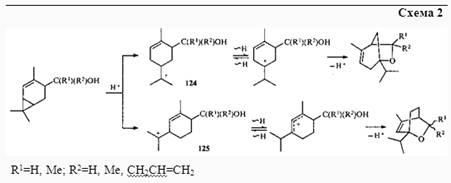
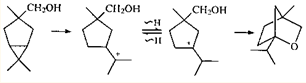
Весьма интересны превращения карановых структур под действием суперкислот. В суперкислых средах увеличивается время жизни карбокатионов, что делает возможным их более глубокие вторичные превращения и вовлекает в перегруппировки все возможные реакционные центры: ЦПК. Двойные связи (если они есть), функциональные группы. Одна из таких возможностей — сигматропные сдвиги водорода или алкильных групп. При этом образуются новые катионы, каждый из которых имеет собственную судьбу. Естественно, что разнообразие интермедиатов, участвующих в этих перегруппировках, обеспечивает и разнообразие продуктов реакции.
При взаимодействии спирта 36
и его производных с суперкислотной системой HSO3
F—SO2
FCl была получена смесь продуктов, образование которых объясняется сигматропными сдвигами в первоначально возникающих ионах 124
и 125
(схема 2
).36
Подобным образом протекает перегруппировка изомерного спирта 90
.36

Для осуществления циклизации такого типа требуется 1,2-водородный сдвиг в п
-ментеновом ионе 126
, так как атом кислорода гидроксиметильной группы удален от атома С(8), на котором возникает первоначальный заряд. В том случае, когда атом кислорода гидроксиметильной группы находится ближе к атому С(8), циклизация происходит через первоначально образующийся м
-ментеновый ион 127
. Конечно, 1,2-сдвиг происходит и в ионе 127
, но атомы кислорода и углерода С(3) (на котором после сдвига сосредоточен положительный заряд) слишком сближены, поэтому циклизация не может осуществиться из-за стерического напряжения.
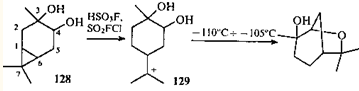
Циклизация диола 128
происходит без предварительного водородного сдвига через п
-ментановый ион 129
, в котором атом кислорода находится ближе к заряженному атому углерода, чем в ионе 126
. При этом происходит селективное раскрытие циклопропанового кольца по связи С(1)—С(7); причина такой селективности пока не выяснена.36
Трициклический спирт 130
перегруппировывается в суперкислотной среде с раскрытием связи С(4)—С(5) с образованием соединения 131
. Циклизация осуществляется за счет 1,2-сдвига водорода в катионе А.36

Перегруппировка спирта 130
примечательна тем, что его молекула имеет два трехчленных цикла, но под действием суперкислоты раскрывается только один из них. Это может быть связано с разницей энергий ВЗМО обоих циклопропановых фрагментов, имеющих различные заместители, хотя электростатическое влияние гидроксиметильиой группы на процесс, который контролируется зарядом, кажется неожиданно большим.
Перегруппировки, сопровождающиеся циклизацией, осуществляются в суперкислотных средах также для секокарановых структур.36
В этом случае положительный заряд должен быть сближен с кислородным атом, чтобы между ними образовалась связь.
При перегруппировках α-транс
-3,4-эпоксикарана*
(75
) в суперкислотных средах в промежуточно образовавшихся ионах происходят не только водородные, но и алкильные сдвиги, и именно они ответственны за перестройку каранового скелета.117
В ионе 4-гидроксикарана 132
должен произойти 1,2-алкильный сдвиг, вызывающий сужение шестичленного цикла с образованием иона 133
; выброс протона и ионизация ЦПК создают возможность циклизации (путь а
).
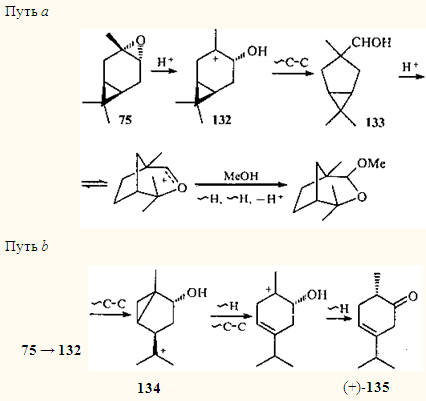
Более интересен другой путь преобразования иона 132
, формально происходящий как. 1,3-алкильный сдвиг и приводящий к иону 134
(путь b
). Однако в заряженных системах не могут осуществляться 1,3-алкильные сдвиги: на атоме С(2) отсутствует подходящая p
-орбиталь. Таким образом, в действительности происходит 1,2-алкильный сдвиг, но в образовании ПС участвуют атомы углерода, не являющиеся соседними (С(1) и С(3)). Перекрывание происходит не вдоль σ-связи, а через пространство; такой сдвиг можно назвать гомоенильным. В конечном итоге это приводит к замыканию трехчленного цикла и иону 134
(β-циклопропильная перегруппировка 118
), в котором происходят последовательные водородный и алкильный сдвиги с образованием конечного кетона (+)-135
.117
Для объяснения перегруппировок β-цис
-3,4-эпоксикарана (136
) в суперкислотной среде предложен механизм с участием бициклического иона 137
, изомерного иону 134
.118
В этом случае стереоспецифично образуется кетон (—)-135
, который является не только стереоизомером, но и энантиомером кетона (+)-135
, получающегося в процессе перегруппировок эпоксида 75
.
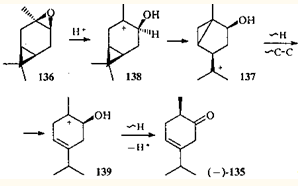
Различную реакционную способность эпоксидов 75
и 136
авторы 118
объясняют термодинамическим и орбитальным факторами: ионы 132
и 138
имеют одинаково благоприятную для процесса сужения цикла ориентацию мигрирующей связи и вакантной орбитали. Для иона 132
энергетически более выгодным является процесс сужения цикла, а для иона 138
предпочтительна β-циклопронильная перегруппировка (через катионы 137
и 139
), поскольку ориентация вакантной орбитали в этом катионе более благоприятна для перегруппировки, чем в ионе 132
.
Приведенные выше схемы превращений эпоксидов 75
и 136
рассматривают лишь наиболее вероятные пути образования конечных продуктов. Соединения, имеющие углеродный скелет 1-метил-4-изопропилбицикло[3.1.0]гексана, которые могли бы получаться из ионов 134
и 137
, среди продуктов превращения эпоксикаранов в суперкислотной среде не найдены. Однако в 1962 г, при кислотной гидратации цис
-эпоксида 136
был получен диол 140
с таким же скелетом,119
и в 1972 г. была подтверждена его структура.120

В 1966 г. было показано, что одним из продуктов дегидратации моноацетата 3β, 4α-карандиола 141
является ацетат транс
-1-метил-4-изопропенилбицикло[3.1.0]гексан-2-ола (142
).121
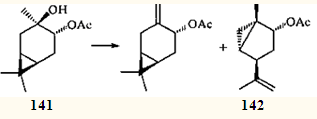
Кропп, исследовавший эту реакцию,121
считая ее первым документально подтвержденным примером перегруппировки с трансаннулярным циклопропильным участием.10
Он полагал, что в реакциях нуклеофильного замещения ЦПК выступает в роли внутреннего нуклеофила и в подобных реакциях чрезвычайно велика роль стереохимического фактора: диметилциклопропановое кольцо должно иметь пространственную возможность атаковать атом С(3), несущий β-заместитель. Важно также наличие α-заместителя у атома С(4) для того, чтобы фиксировать шестичленный цикл в конформации "син
-ванны" А
.121

Относительно высказанных Кроппом стереохимических условий "циклопропильного участия" существовали разные мнения. Предложенная им интерпретация пространственных особенностей протекания подобных реакций какое-то время считалась общепринятой и цитировалась в специальной литературе.12
Однако последующие исследования 10, 122-128
позволили выдвинуть и экспериментально обосновать предположение, что в переходном состоянии молекулы реагирующих карановых соединений имеют конформацию "анти
-ванны" (B-D
); ориентация заместителя при С(4) значения не имеет.122, 123, 129
Фактически "трансаннулярное участие ЦПК" сводится к 1,2-гомоенильному сдвигу, который может произойти (и происходит) в заряженных системах. Отсутствие продуктов "трансаннулярного циклопропильного участия" при перегруппировках в суперкислотах обусловлено тем, что вследствие чрезвычайно малой нуклеофильности среды и низких температур ионные процессы идут глубже и отличаются высокой селективностью. Поэтому в суперкислотах превращения не останавливаются на ионе 137
, а идут дальше с образованием иона 139
. который и приводит к конечному продукту реакции.

Такой подход объясняет общность превращений эпоксида 136
в условиях обычного кислотного катализа и в суперкислотах, но при этом возникают новые вопросы. Превращение зпоксида 75
в суперкислотах приводит к продуктам, предполагающим промежуточное образование иона 134
, а сольволиз соединения 75
протекает без образования 1-метил-4-изопропилбицикло[3.1.0]гептановых структур. В качестве побочного продукта процесса происходит сужение цикла в результате алкильного сдвига в ионе 132
, что приводит к иону 133
, который стабилизируется путем выброса протона; в суперкислоте ион 133
подвергается перегруппировке.125, 130

Несомненно, эпоксиды 75
и 136
реагируют в обычных условиях по-разному вследствие различий в их пространственном строении. Однако в суперкислотах эти различия по отношению к "трансаннулярному циклопропильному участию" не проявляются или, по крайней мере, не играют существенной роли. Можно предположить, что в суперкислотах происходит более глубокое разделение зарядов, что меняет геометрию каранового скелета и создает возможность 1,2-сдвига. В обычных кислотах на участвующих атомах сконцентрирован лишь частичный заряд,121
поэтому геометрия каранового скелета изменяется незначительно. Следует, однако, отметить, что ион 133
образуется в условиях обычного кислотного катализа, что пока необъяснимо с точки зрения приведенной выше аргументации.
При взаимодействии α-эпоксикарана с альдегидами на глине асканит-бентонит (алюмосиликатный катализатор) не образуется продуктов, которые указывали бы на участие в реакции иона 134
.131

В этом случае глубокие превращения ионов не происходят, так как альдегид перехватывает ион 132
и реакция идет по другому пути. В то же время эпоксид 136
в аналогичных условиях дает продукты, которые свидетельствуют о том, что ион 138
претерпевает не только 1,2-гомоенильный, но и 1,2-алкильный сдвиг с образованием иона 133
(что не наблюдалось в суперкислотах 118
), который затем перегруппировывается.
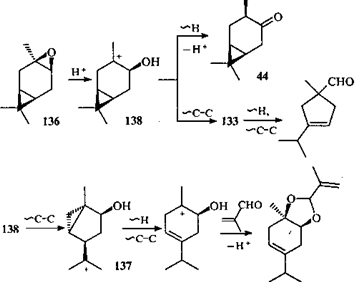
Специфика каталитических превращений на твердой поверхности приводит к тому, что из эпоксида 136
не образуется ацеталь с карановым скелетом, но получается кетон 44
, который отсутствует в реакции эпоксида 75
.
На глине асканит-бентонит 3-карен в реакции с альдегидами не вступает,131
но спирт 36
в этих условиях дает целый ряд перегруппированных продуктов, строение которых зависит от использованного альдегида.132


Предложенный авторами132
механизм реакции включает атаку молекулы протонированного масляного альдегида по более замещенному концу двойной связи и образование не третичного, а менее устойчивого вторичного катиона 143
. Его перегруппировка с раскрытием ЦПК дает ионы 144, 145
и далее продукты реакции. В разделе II
.1 приводится другая точка зрения на образование продуктов с похожим скелетом.24, 25
В пользу механизма, предложенного в работе 132
, свидетельствует получение трициклических продуктов в реакции спирта 36
с α-метилакролеином (схема 3
).
Близость двойной связи и катионного центра в ионе 146
может привести к циклизации, которая, как полагают, обусловлена наличием метильной группы в α-положении к двойной связи.
Выше, на примерах эпоксикаранов, уже описывались перегруппировки каранового скелета в бицикло[3.1.0]гексановый. Похожая (с формальной точки зрения) перестройка каранового скелета наблюдается в процессе превращения оксима кар-2-ен-4-она (147
) под действием азотистой кислоты.133
С учетом природы функционального заместителя наиболее вероятный путь превращения оксима 147
включает интермедиаты 148 - 152
.
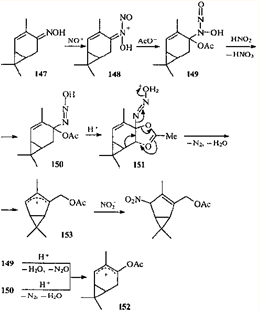
Очевидно, что за перестройку каранового скелета ответственен сдвиг алкильной группы в заряженной системе. Проведя квантово-химические расчеты, авторы 133
пришли к заключению, что 1,2-алкильный сдвиг в аллильном ионе 152
невозможен с точки зрения его пространственного строения и термодинамики. Что касается иона 151
, то в нем предполагается согласованное элиминирование азота с одновременной атакой ацетатной группы и миграцией связи С(5)—С(6). Катион 153
стабилизируется захватом аниона NO2
¯
. Переходное состояние типа 151
постулировалось ранее130, 134
для объяснения пиролиза моноацетата карандиола 141
.
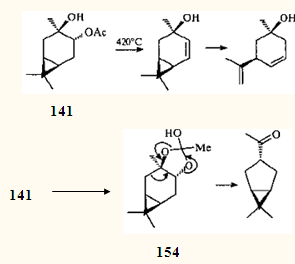
Однако предложенный авторами циклический интермедиат 154
лишь формально - схематически описывает перестройку связей. Очевидно, что термический сдвиг связи С(2)—С(3) может произойти только в ионе. Возможно, уже отщепившаяся (но согласованному механизму) и присутствующая в реакции уксусная кислота способствует образованию промежуточных ионов.
Еще более удивительный пример скелетной перегруппировки, обусловленной участием оксииминной группы, описан в работе 135
. Реакция оксимов 3α-гидроксиламинокаран-4-она (155а
) и 3α-(O
-ацетил)гидроксиламинокаран-4-она (155b
) с боргидридом натрия в ацетонитриле приводит к продукту реакции 156
с семичленным конденсированным циклом.
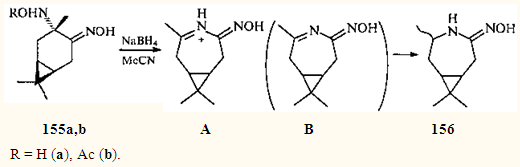
В данном случае ацетонитрил, по-видимому, является не только растворителем, но и реагентом, так как и его отсутствие перегруппировка не происходит. АвторыI
постулируют два возможных интермедиата реакции — А
или В
, — но отмечают, что механизм их образования пока неясен.
V
.Выводы
Рассмотренный в курсовой работе обширный материал свидетельствует, что изучение химических превращений карановых производных выходит далеко за рамки того раздела органической химии, который принято называть химией терпеноидов. 3-Карен — поистине уникальное природное соединение, предоставляющее исследователям огромные возможности. Благодаря доступности 3-карена, из него сравнительно легко получают все новые производные, которые представляют интерес как сами по себе, так и служат прекрасными моделями для изучения механизмов реакций. По нашему мнению, среди природных соединений только бензол может сравниться с 3-кареном по вкладу в теоретическую и синтетическую органическую химию.
Хотя мы постарались рассмотреть механизмы известных перегруппировок, внимательный читатель, очевидно, отметил, что они часто носят гипотетический, а иногда и спорный характер. Нет сомнений, что исследования в этой области будут продолжены и не только дадут ответы на сегодняшние вопросы, но и поставят новые. Мы надеемся, что начатая нами аналитическая работа будет продолжена в рамках дипломного проекта.
Литература
1. S.Dev.
Curr
.
Sci
.,
52
, 1125 (1983)
2. B.V.Lawrence. Perfum.Flavor
., 33
(3), 66 (2001)
3. Н.Ф.Салахутдинов, В.А.Бархаш. Успехи химии
, 66
, 376 (1997)
4. H.R.Sonawane, B.S.Nanjundiah, M.U.Kumar. Tetrahedron Lett
., 25
,2245(1984)
5. A.S.Khanra.K.K.Chakravarti, R.B.Mitra. Indian J. Cliem
., 13
,314 (1975)
6. А.Х.Хусид, О.М.Нефедов. Журн. Всесоюз. хим. о-ва им. Д.И.Менделеева,
33
, 653 (1988)
7. С.А.Осадчий, Г.А.Толстиков. Химия в интересах устойчивого развития,
5
,79 (1997)
8. H.Sadowska, J.Gora. Perfum. Flavor
., 7
(1), 52 (1982)
9. J.Verghese. Perfum. Flavor
., 4
(4), 23 (1979)
10. Б.А.Арбузов, З.Г.Исаева. Успехи химии
, 45
, 1339(1976)
11. C.P.Mathew, K.K.Sugathan, J.Verghese. J. Sci. Ind. Res., 22
, 173 (1963)
12. W.F.Erman. In Studies in Organic Chemistry. Vol. 11.
(Ed. P.Gassman). Marcel Dekker, New York, 1985. Pts A, B
13. D.H.Grayson. Nat. Prod. Rep
., 15
,439 (1998)
14. D.H.Grayson. Nat.
Prod. Rep
.. 17
, 383 (2000)
15. Н.Ф.Салахутдинов. Химия в интересах устойчивого развития
,5
,21(1997)
16. В.А.Бархаш, М.П.Половинка. Успехи химии
, 68
, 430 (1999)
17. В.А.Чуйко, Э.Н.Мануков, Ю.В.Чижов, М.М.Тимошенко. Химия природ. соединений
, 639 (1985)
18. И.И.Бардышев, Э.Н.Мануков. Журн. орг. химии
, 1
, 1426 (1965)
19. A.D.Walsh. Trans. Faraday Soc.
, 45
, 179 (1949)
20. A.de Meijere. Angew. Chem., Int. Ed. Engl
., 18
, 809 (1979)
21. W.Cocker, P.V.R.Shannon, P.A.Staniland. J. Chem. Soc. C
, 915 (1967)
22. W.Cocker, D.P.Hanna, P.V.R.Shannon. J. Chem. Soc. C
, 489 (1968)
23. W.Cocker, D.P.Hanna, P.V.R.Shannon. J.
Chem. Soc. C
, 1302 (1969)
24. J.Chlebicki, B.Burczuk. Tetrahedron Lett.,
4775 (1970)
25. J.Chlebicki, B.Burczuk. Rocz.
Chem
.,45
, 1225(1971)
26. K.Gollnick, G.Schade, S.Schroeter. Tetrahedron
, 22
, 139 (1966)
27. K.Gollnick, G.Schade. Tetrahedron Lett.,
2335 (1966)
28. А.А.Фокин, Е.Д.Бутова, И.В.Коломицин, Е.А.Гагаева, И.В.Гогоман, А.М.Корнилов, А.Е.Сорочинский, А.Г.Юрченко, П.А.Красуцкий. Журн
.
орг
.
химии
, 30
, 669 (1994)
29. В.А.Чуйко.Э.Н.Мануков, Н.Г.Яремченко. Весц
i
АН
Белару
ci
. Сер
.
х
i
м
.
н
a
вук
, (3), 70 (1996)
30. A.R.Rossi. J. Phys.
Chem
., 83
, 2554 (1979)
31. F.H.Allen. Acta Crystullogr., Sect.
B
, 36
, 81 (1980)
32. О.Г.Выглазов, Э.Н.Мануков, В.А.Чуйко, Т.Р.Урбанович.Журн
.
орг
.
химии
, 28
, 968 (1992)
33. W.Cocker, H.St.J.Lauder, P.V.R.Shannon. J. Chem. Soc.. Perkin Trans
. 1
,332(1975)
34. Г.А.Толстиков, А.Ю.Спивак, Л.М.Халилов, Е.В.Васильева, С.И.Ломакина, И.А.Иванова. Изв
.
АН
СССР
.
Сер
.
хим
., 1814 (1985)
35. Э.Н.Мануков, О.Г.Выглазов, В.А.Чуйко, Е.С.Мардилович. Журн
.
орг
.
химии
, 24
, 1449 (1988)
36. М.П.Половинка, О.Г.Выглазов, Д.В.Корчагина, Э.Н.Мануков, В.А.Бархаш. Журн
.
орг
.
химии
,
28
, 2253 (1992)
37. В.А.Чуйко, О.Г.Выглазов, Л.Ю.Тычинская. Журн
.
орг
.
химии
, 34
,1357(1998)
38. T.Norin, S.Strombcrg, M.Weber. Chem. Scr
., 20
, 49 (1982)
39. H.R.Sonawane, B.S.Nanjundiah, P.C.Purohit. Tetrahedron Lett.,
24
,3917(1983)
40. W.J.Leigh, R.Srinivasan. J. Am. Chem. Soc.,
105
, 514 (1983)
41. H.Hart, T.Takino. J. Am. Chem. Soc
., 93
, 720 (1971)
42. P.J.Kropp. J. Am. Chem. Soc
., 89
, 1126 (1967)
43. H.R.Sonawane, V.G.Naik, B.S.Nanjundiah, P.C.Purohit. Tetrahedron Lett.,
24
, 3025 (1983)
44. D.C.Heckert, P.J.Kropp. J. Am. Chem. Soc.,
90
, 4911 (1968)
45. G.Buchi, E.M.Burgess. J. Am. Chem. Soc.,
82
, 4333 (1960)
46. R.K.Murray Jr.,, T.M.Ford. J. Am.
Chem. Soc.,
102
, 3194 (1980)
47. J.A.Bcrson, M.R.Willcott III. J. Am.
Chem. Soc
., 88
, 2494 (1966)
48. E.Ciganec. J. Am. Chem. Soc.,
87
, 652 (1965)
49. G.Maier. Angew. Chem.,
79,
446 (1967)
50. F.-G. Klarner. Tetrahedron Lett
., 19
(1974)
51. G.Maas, M. Regitz. Chem. Ber.,
109
, 2039 (1976)
52. R.Neidlcin, C.M.Radke. Helv. Chim. Acta,
66
, 2626 (1983)
53. R.Hoffmann. Tetrahedron Lett
., 2907 (1970)
54. M.Gorlitz, H.Gunther. Tetrahedron
, 25
, 4467 (1969)
55. E.Ciganec. J. Am. Chem. Soc.,
93
, 2207 (1971)
56. R.B.Woodward, R-HofTmann. Angew. Chem
., 81
, 797 (1969)
57. M.B.Rubin. J. Am. Chem. Soc
., 103
, 7791 (1981)
58. И.И.Бардышсв, Г.В.Дещиц, А.А.Вахрамеева. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
,
(2), 69 (1980)
59. A.Baeyer. Berichfe
, 27
, 810 (1894)
60. T.Saecd, P.J.Sondra, M.J.E.Verzcle. Phytochemistry
, 17
, 1433 (1978)
61. E.J.Corey, H.J.Burke. J. Am. Chem. Soc
., 78
, 174 (1956)
62. E.J.Corey, H.J.Burke, W.A.Remers. J. Am.
Chem. Soc.,
78
, 180 (1956)
63. W.D.P.Burns, M.S.Carson, W.Cocker, P.W.R.Shannon. J. Chem Soc.C
, 3073 (1968)
64. О.Г.Выглазов, Э.Н.Мануков, Б.Г.Ударов, Г.Н.Бажина, Т.Р.Урбанович, Л.В.Изотова. Химия природ, соединений
, 289 (1989)
65. О.Г.Выглазов, Э.Н.Мануков, М.Н.Федорищева, Н.Г.Арико, В.А.Чуйко, Г.Н.Бажина. Химия природ, соединений
, 328 (1991)
66. L.Ruzicka. Pure Appl. Chem.,
6
, 493 (1963)
67. Г.А.Рудаков, Ю.А.Подтавченко, Л.С Иванова. В кн. Синтетические продукты из канифоли и скипидара.
Волго-Вятское книжное изд-во, Горький, 1970. С. 156
68. Э.Н.Мануков, В.А.Чуйко, П.В.Кузьмичкин. Химия природ. соединений
, 783(1979)
69. Э.Н.Мануков, В.А.Чуйко. Химия природ, соединений
, 450 (1983)
70. W.Cocker, P.V.R.Shannon, P.A.Staniland. J. Chem. Soc.
C
, 41
(1966)
71. G.O.Schenck. Angew. Chem
., 69
, 579 (1957)
72. Э.Н.Мануков, В.А.Чуйко. Химия
древесины
, (4), 48 (1983)
73. S.M.Davis, G.A.Somorjai. J. Calal.,
65
, 78 (1980)
74. Э.Н.Мануков, В.А.Чуйко, О.Г.Выглазов. Химия природ. соединений,
259 (1982)
75. Э.Н.Мануков, В.А.Чуйко, О.Г.Выглазов. Журн. орг. химии
, 20
, 289(1984)
76. Э.Н.Мануков, В.А.Чуйко, Е.С.Мардилович, О.Г.Выглазов. Весцi АН Беларуci. Сер. хiм. нaвук
, (3), 55 (1988)
77. Э.Н.Мануков, Г.Н.Бажина. Журн. орг. химии
, 24
, 121 (1988)
78. Э.Н.Мапуков, В.А.Чуйко, О.Г.Выглазов. Журн. орг. химии
, 19
, 662(1983)
79. Э.Н.Мапуков, О.Г.Выглазов, В.А.Чуйко, И.А.Шингсль. Журн.
орг
. химии
, 21
, 2089 (1985)
80. О.Г.Выглазов, В.А.Чуйко, Э.Н.Мапуков. Весцi
АН
Беларуci
. Сер.
хiм
.
нaвук
, (6), 40 (1988)
81. Э.Н.Мануков, О.Г.Выглазов, И.А.Шингель. Весц
i
АН Беларуci.
Сер.
хiм
.
нaвук
, (1), 67 (1986)
82. Э.Н.Мануков, О.Г.Выглазов. Химия природ., соединений
, 534 (1983)
83. G.Ohloff, H.Farnow, W.Phillipp. Liehigs Ann. Chem
., 613
, 43 (1958)
84. О.Г.Выглазов, Э.Н.Мануков, В.А.Чуйко, Б.Г.Ударов. Весц
i
АН
Белару
ci.
Сер. хiм. нaвук
, (3), 42 (1991)
85. Т.Джилкрист, Р.Сторр. Органические реакции и орбитальная симметрия
. Мир, Москва, 1976
86. Э.Н.Мапуков, Т.Р.Урбанович, О.Г.Выглазов, В.А.Чуйко, Е.Д.Скаковский. Химия природ. соединений,
193 (1989)
87. D.S.Glass, R.S.Boikcss, S.Winstcin. Tetrahedron Lett
., 999 (1966)
88. C.A.Coulson,W.E.Moffitt. Phylos. Mag.,
40
, 1 (1949)
89. M.C.Flowers, H.M.Frcy. J. Chem. Soc.,
3547 (1961)
90. G.Ohloff. Chem. Ber
., 93
, 2673 (1960)
91. G.Ohloff. Tetrahedron Left.,
3795 (1965)
92. K.Gollnik. Tetrahedron Lett
., 327 (1966)
93. R.J.Loncharich, K.N.Houk. J. Am. Chem. Soc.,
110
, 2089 (1988)
94. О.Г.Выглазов, С.Л.Быховсц, Э.Н.Мануков, Т.Р.Урбаиович. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
,
(2), 65 (1991)
95. W.von Doering, E.K.G.Schmidt. Tetrahedron
, 27
, 2005 (1971)
96. L.L.Frolova, A.V.Kutchin. In International Conference on Natural Products and Physiologically Active Substances. (Abstracts of Reports)
.Novosibirsk, 1988. P. 109
97. В.А.Чуйко, Э.Н.Мануков, О.Г.Выглазов. Докл
.
АН
БССР
. 30
, 158(1986)
98. В.А.Миронов, А.Д.Фeдорович, А.А.Ахрем. Успехи
химии
, 50
, 1272(1981)
99. W.J.Bouma, M.A.Winecnt, L.Radom. Int. J. Quantum Chem
., 14
, 767(1978)
100. J.A.Berson, L.Salem. J. Am. Chem. Soc
., 94
. 8917 (1972)
101. A.P.ter Borg, H.Kloosterziel, N.van Meurs. Proc. Chem. Soc., London
, 359 (1962)
102. F.-G.Klarner. Angew. Chem.. Int. Ed. Engl.,
13
, 268 (1974)
103. F.-G.Klarner, S.Yaslak, M.Wcttc. Chem. Ber.,
112
, 1168 (1979)
104. F.-G.Klarner, B.Brassel. J. Am. Chem. Soc.,
102
, 2469 (1980)
105. A.Jarzecki, J.Gajewski, E.R.Davidson. J. Am. Chem. Soc
., 121
, 6928(1999)
106. A.Kless, M.Nendel, S.Wilsey, K.N.Houk. J. Am. Chem. Soc
., 121
, 4524(1999)
107. Э.Н.Мануков, О.Г.Выглазов, В.А.Чуйко, Е.Д.Скаковский. Журн
.
орг
.
химии
, 22
, 1105 (1986)
108. Э.Н.Мануков, О.Г.Выглазов, В.А.Чуйко. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
, (5), 58 (1989)
109. Э.Н.Мануков, О.Г.Выглазов, Б.Г.Ударов. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
, (4), 51(1990)
110. M.Nendel, B.Goldfuss, B.Beno, K.N.Houk, K.Hafner, H.-Y.Lindner. Pure Appl. Chein
., 71
, 221 (1999)
111. D.A.Modarelli. J. Org. Chem
., 65
, 7277 (2000)
112. О.Г.Выглазов, Э.Н.Мануков. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
,(5), 111(1988)
113. Г.Н.Бажипа, В.А.Чуйко, Э.Н.Мануков. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
, (4), 47 (1993)
114. О.Г.Выглазов, Э.Н.Мануков, В.А.Чуйко. Журн
.
орг
.
химии
, 25
, 1571 (1989)
115. В.А.Чуйко, О.Г.Выглазов, Э.Н.Мануков, Л.Ю.Тычинская. Весц
i
АН
Белару
ci.
Сер
.
х
i
м
.
н
a
вук
,
(2), 54 (1998)
116. W.von Docring, W.R.Roth. Tetrahedron
. 19
, 715 (1963)
117. М.П.Половинка, О.Г.Выглазов, Д.В.Корчагина, В.А.Бархат. Журн
.
орг
.
химии
, 27
, 2623 (1991)
118. М.П.Половинка, Д.В.Корчагина, Ю.В.Гатилов, О.Г.Выглазов, Г.А.Зенковец, В.А.Бархаш. Журн
.
орг
.
химии
, 34
, 1342 (1998)
119. Z.Chabudzinski, H.Kuczynski. Roc:. Chem.,
36
, 1173 (1962)
120. Б.А.Арбузов, В.А.Шайхутдннов, З.Г.Исаева. Изв
.
АН
СССР
.
Сер
.
хим
.
, 2124(1972)
121. P.J.Kropp..J
. Am. Chem. Soc.,
88
, 4926 (1966)
122. Б.А.Арбузов, З.Г.Исаева, Р.Р.Дьяконова. Изв
.
АН СССР. Сер. хим.,
2778 (1980)
123. Б.А.Арбузов, З.Г.Исаева, Р.Р.Дьяконова. Изв.
АН СССР. Сер.хим.,
2141 (1980)
124. Б.А.Арбузов, З.Г.Исаева, Р.Р.Дьяконова. Изв. АН СССР. Сер. хим.,
972 (1975)
125. Б.А.Арбузов, З.Г.Исаева, Э.Х.Казакова. Докл. АН СССР
, 215
, 113(1974)
126. H.Kuczynski, M.Walkowicz, C.Walkowicz, K.Nowak, I.Sicmion. Rocz. Chem.,
38
, 1625 (1964)
127. M.Walkowicz, H.Kuczynski. Rocz. Ch
e
m.,
40
, 1231 (1966)
128. E.Maslinski, E.Michalek. Roc
z
. Chem
., 47
, 285 (1973)
129. Р.Р.Дьяконова, А.А.Мусина, Р.Г.Гайнуллина, П.П.Чернов. Изв. АН СССР. Сер. хим
., 1327 (1982)
130. J.C.Leningwell, R.E.Shackelford, H.J.Young. Synth. Comimm
., 105(1972)
131. К.П.Волчо, Л.Е.Татарова, Д.В.Корчагина, Н.Ф.Салахутдинов, В.А.Бархаш. Журн. орг. химии,
36
, 41 (2000)
132. И.В.Ильина, К.П.Волчо, Д.В.Корчагина, Н.Ф.Салахутдинов, В.А.Бархаш. Журн. орг. химии,
35
, 699 (1999)
133. A.V.Tkachev, A.M.Chibiryaev, A.Yu.Denisov, Yu.V.Gatilov. Tetrahedron
.51
, 1789(1995)
134. J.C.Lemngwell, R.E.Shackelford. Tetrahedron Lett.,
2003 (1970)
135. P.A.Petukhov, A.Yu.Denisov, A.V.Tkachev. Tetrahedron
, 53,
2527 (1997)
*
Символами α
и β
в химии терпеноидов традиционно указывают пространственное расположение функциональных групп производных карана: α
-заместители и трехчленный цикл расположены по разные стороны циклогексанового кольца (транс
). β-заместители и ЦПК — по одну сторону (цис
).
|