Содержание
Ведение
1. Нуклеофильное замещение галогена в молекуле органического соединения
1.1 Основные сведения о механизме реакции
1.2 Основные факторы, влияющие на ход процесса нуклеофильного замещения
1.3 Замена атома галогена на - ОН, - ОR, - OН, - SН и -SRгруппы
1.4 Замена атома галогена на группы -NН2
,-NНR,-NR2
1.5 Замена атома галогена на -СN и -SO3
Na
2. Нуклеиновое замещение сульфогруппы
2.1 Общие сведения о процессе
2.2 Примеры осуществления нуклеофильной замены сульфогруппы в промышленности
3. Особенности техники безопасности при проведении процессов нуклеофильной замены галогена и сульфогруппы
Заключение
Список использованных источников
Введение
Атом галогена в молекуле органического соединения с успехом может быть замещен на другие группы атомов, что создает широкие возможности для синтеза биологически активных соединений, исходя из галогенпроизводных. Так, на основе галогензамещенных могут быть получены амины, спирты, фенолы, эфиры, тиолы, сульфиды, алкилнитриты и нитроалканы, нитрилы и изонитрилы, лкены, алкены и др. соединения.
Реакции нуклеофильного замещения у насыщенного атома углерода являются одной из наиболее изученных в органической химии с точки зрения механизма. Основной вклад в установление в установление механизма внесён английской школой химиков во главе с К. Ингольдом. Уже в 30-х годах ХХ столетия на основе фундаментальных исследований кинетики и стереохимии было представлено два предельных механизма нуклеофильного замещения – бимолекулярное нуклеофильное замещение SN
2 и мономолекулярное замещение SN
1.
Реакции нуклеофильного замещения галогена и сульфогруппы являются весьма важными и распространенными в органическом синтезе и синтезе лекарственных веществ, а так же витаминов. [1]
1. Нуклеофильное замещение галогена в молекуле органического соединения
1.1 Основные сведения о механизме реакции
Нуклеофильное замещение в ряду галогеналканов может осуществляться как по SN
1, так и по SN
2 механизмам. В первом случае лимитирующей скорость процесса стадией является диссоциация галогеналкана по связи С - Hal с образованием карбкатиона:
 (1) (1)
Таким образом, скорость процесса в этом случае не должна зависеть от концентрации нуклеофила. При достаточно большом времени существования иона карбения для оптически активных галогеналканов должна наблюдаться рацемизация. При соизмеримых величинах констант скоростей последовательных стадий процесса одна сторона катиона будет экранирована сольватированным галогенид-анионом и атака нуклеофила будет более вероятна с другой стороны, что приведет к преимущественному обращению конфигурации. Однако основной причиной отсутствия полной рацемизации является то, что во многих случаях продукты реакции образуются не из свободных карбкатионов, а из ионных пар.
 (2) (2)
Молекула исходного соединения может диссоциировать с образованием контактной ионной пары (а), сольватно – разделённой ионной пары (b) и диссоциированных сольватирорванных ионов (с). В контактной ионной паре ассиметрия в значительной мере сохраняется, а потому нуклеофильная атака приводит к обращению конфигурации. При атаке сольватно – разделённой ионной пары селективность снижается, что приводит к общей рецемизации. Если образуется свободный радикал, то рецемизация должна быть полной. Обычно обращение конфигурации при механизме SN
1 составляет от 5 до 20%.
В случае молекулярного замещения может протекать ряд побочных процессов, протекающих через стадию образования иона карбения, в частности элиминирование (Е1):
 (3) (3)
Бимолекулярное замещение SN
2 обычно происходит как синхронный процесс:
 (4) (4)
При этом механизме нуклеофил Y приближается к субстрату со стороны, противоположной проходящей группе. Реакция представляет собой одностадийный процесс, в котором промежуточное соединение не образуется. Связь С-Y образуется одновременно с разрывом связи С-Х. В переходном состоянии исходная sp3
-гибридизацию с примерно перпендикулярной р-орбиталью. Одна доля этой р-орбитали перекрывается с нуклеофилом, а вторая с уходящей группой. Поэтому механизм SN
2, В котором происходила бы фронтальная атака, иногда не наблюдается.
При таком механизме скорость процесса существенно зависит как от природы, так и от концентрации нуклеофила. Реакция всегда сопровождается обращением конфигурации. Побочной может быть реакция Е2. [2]
Необходимо помнить о том, что термины SN
1 и SN
2 указвают лишь на молекулярность, но не на порядок реакции. Поэтому скорость реакции, протекающей по механизму SN
2, будет отвечать уравнению второго порядка лишь в случае, когда оба компонента находятся в малых контролируемых концентрациях. При большом избытке нуклеофильного агента реакция может протекать по первому или дробному. Аналогичное положение может создаться, когда оба компонента не являются кинетически независимыми (например, при образовании ионных пар при неполярных растворителях). Ингольд отмечает, что и для реакции SN
1 не всегда характерен первый порядок.
Очевидно, что влияние полярности среды на скорость и механизм процесса будет достаточно сильным.
Тип механизма (SN
1 или SN
2) зависит от структуры реагирующих соединений. Природа галогена обычно мало влияет на механизм реакции, но значительно изменяет ее скорость. С увеличением разветвленности R начинает преобладать механизм SN
1, так как при этом создаются стерические препятствия для прохождения реакции по механизму SN
2 и увеличивается стабильность промежуточного карбкатиона. Чем выше нуклеофильность реагента, тем вероятнее механизм SN
2. [1]
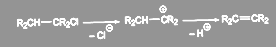
Аллилгалогениды легко вступают в реакции нуклеофильного замещения. В условиях, благоприятных для протекания мономолекулярных реакций, образуется смесь двух соединений, так как промежуточный мезомерный аллилкатион может в зависимости от условий приводить к двум разным продуктам:
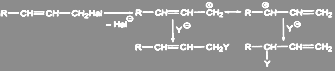 (5) (5)
При механизме SN
2 перегруппировка не происходит:
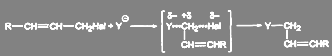 (6) (6)
Если подход сильного нуклеофила к атому углерода при галогене стерически затруднен, то в неполярном растворителе реакция может идти с аллильной перегруппировкой при сохранении механизма SN
2:
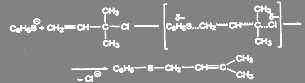 (7) (7)
В ароматическом ряду (галоген в ядре) замещение идёт значительно труднее.
По мономолекулярному механизму реакция протекает лишь в исключительных случаях. Примером может служить получение гидроксисоединений из солей диазония:
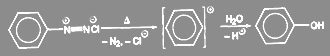 (8) (8)
Обычно нуклеофильная замена галогена в ароматическом ядре протекает по биомолекулярному механизму SN
Ar. В отличие от описанного для алкилгалогенидов реакция идёт не по синхронному механизму, так как атакующий нуклеофил способен образовывать новую связь раньше, чем порвётся старая, и первая стадия обычно определяет скорость всей реакции:
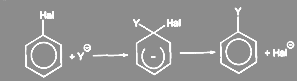 (9) (9)
Существование таких отрицательно заряженных σ-комплексов было доказано экспериментально:
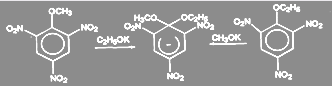 (10) (10)
Подобные интермедиаты представляют собой устойчивые соли, называемые солями Мейзенгеймера, со времени обнаружения их в 1902 г было выделено большое число таких солей, строение нескольких интермедиантов такого типа было подтверждено данными ЯМР и pентгеноструктурного анализа. Однако описанный механизм не является единственно возможным. С помощью меченого атома углерода было показано что в арилгалогенидах, не содержащих активирующих групп, замещающая группа становится не только к тому атому углерода, где был галоген, но в равной степени и к соседнему атому:
 (11) (11)
Идентичность соседних положений при отсутствии других заместителей в ядре объясняется тем, что реакция идет по механизму отщепления-присоединения (кинезамещения) через стадию образования 1,2-дегидродензола:
 (12) (12)
Промежуточное образование дегидробензола было доказано как физико-химическими, так и чисто химическими методами. Так, при действии амальгамы лития на l-фтор-2-бромбензол в присутствии диенофилов промежуточно образующийся 1 ,2-дегидробензол вступает с ними в реакцию Дильса-Альдера:
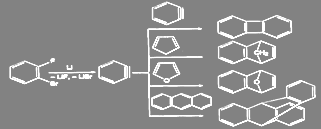
1.2 Основные факторы влияющие на ход процесса нуклеофильного замещения
Условия проведения и ход реакций нуклеофильной замены галогена зависит от многих факторов. При выборе оптимальных условий проведения процесса необходимо учитывать особенности химического строения субстрата и нуклеофильного реагента, полярность среды, природу уходящего галогена.
Относительно связи строения субстрата и его реакционной способности нужно отметить следующее. Скорости SN
1 реакций алкильных производных возрастают в ряду: первичный алкил, вторичный, третичный. Так, константы скоростей реакций гидролиза алкилбромидов при 50 ºС для R-С2
Н5
; (СН3
)2
СН-; (СН3
)3
С- относятся соответственно как 1 : 11,6 : 1,2·106
. Пространственные препятствия в этом случае не имеют большого значения. Более того, увеличение объема заместителей дестабилизирует исходное состояние в большей степени, чем переходное, что должно приводить к увеличению скорости диссоциации. Особенности строения молекулы субстрата, приводящие к стабилизации образующегося карбкатионa, должны приводить к ускорению реакции SN
1 замещения. Это достигается, в частности, при наличии в α-положении к реакционному центру фенильных или аллильных заместителей, а также атомов, имеющих неподелённую пару электронов.
При этом по силе активации один α-фенильный радикал соответствует примерно двум алкильным заместителям. [3]
Что касается влияния строения субстрата на скорость SN
2 замещения то порядок изменения реакционной способности при переходе первичного к третичному радикалу прямо противоположен наблюдаемому при SN
1 замещении. Первичные галогенпроизводные реагируют очень гладко, вторичные - значительно хуже, а третичные часто не реагируют вооще. Пространственные эффекты играют в SN
2 замещении важную роль, и низкая скорость для третичных галогенидов объясняется, в частности пространственными препятствиями для атаки нуклеофилом.
Таким образом, при переходе от первичного алкилгалогенида к тритичному механизм реакции может измениться от бимолекулярного до мономолекулярного. Переход от одного механизма к другому не является резким и зависит от ряда конкретных условий. Принципиально возможно протекание реакции по двум механизмам одновременно.
В ароматических галогенидах, как уже отмечалось, замещение практически всегда происходит по бимолекулярному механизму. Исключением является разложение солей диазония. Влияние других заместителей в ароматическом кольце на легкость замещения галогена изучалось очень широко. Наличие электроноакцепторных заместителей в орто-, пара- положениях существенно облегчает реакцию SN
2 замещения, электродонорных - затрудняет ее.
Сильное ускорение процесса замещения галогена под влиянием орто- и пара-расположенных нитрогрупп хорошо известно. Так, 2,4,6-тринитрогалогенбензолы очень легко реагируют с водой, спиртами, аммиаком, первичными и вторичными аминами, образуя пикриновую кислоту, с эфиры или амиды. Динитрогалогенбензолы реагируют с подобными реагентами медленнее, а мононитро - значительно медленнее. Так, пикрилхлорид гидролизуется так же легко, как хлорангидрид кислоты, замена галогена в о- и п-хлорнитробензоле проходит в щелочном растворе при 130-150 º С, а хлорбензол гидролизуется до фенола лишь при температуре: 350-400 º С и давлении выше 30 МПа под действием 5% раствора щелочи.
В орто- и пара-замещенных хлорбензолах легкость замещения хлора на гидроксил определяется рядом: N02
> > SO3
H > СООН. При этом активация за счет NО2
-группы на несколько порядков выше активации за счет SO3
H и СООН-групп. [10]
При взаимодействии замещенных галогенбензолов с метилатом натрия активирующее действие групп при одинаковом их размещении относительно галогена изменяется в соответствии с рядами:
 (14) (14)
Отмечено значительное увеличение подвижности галогена в ароматическом ядре при наличии в орто- и пара- положении к нему NH3
–группы (т. е. аминогруппы в кислой среде).
Заместители первого рода значительно снижают подвижность галогена и в ряде случаев переводят механизм SN
Аг в кинезамещение через дегидробензол. При переходе к пиридину и хинолину нуклеофильная подвижность галогена повышается. В этом смысле пиридин и хинолин можно рассматривать как аналоги нитробензола. 4-Галогенпиридины активнее 2-замещенных; 3-галогензамещенные еще менее активны и в этом смысле похожи на фенилгалогениды.
При переходе к диазинам нуклеофильная подвижность атома галоген увеличивается. Высокой реакционной способностью выделяются 2- и 4-галогенпиримядины. 2-Хлорпиримидин реагирует с н-бутиламином уже при комнатной температуре, а 4-хлорпиримидин нельзя выделить в индивидуальном состоянии из-за легкого отщепления хлора. 2-Хлорпиразин и 3-хлорпиридазин также значительно активнее 2-хлорпиридина в реакциях нуклеофильного замещения.
В ряду пятичленных гетероциклов реакции нуклеофильного замещения изучены еще недостаточно. Галогензамещенные фураны и тиофены относительно инертны в реакциях нуклеофильного замещения, хотя их реакционная способность выше, чем у соответствующих галогенарилов. Введение сильных электроноакцепторных заместителей увеличивает подвижность галогена.
Пространственные факторы при нуклеофильном замещении в ароматическом ряду не являются определяющими, так как атака направлена сбоку к плоскости ароматического ядра.
В зависимости от природы галогена порядок реакционной способности алкилгалогенидов в реакциях нуклеофильного замещения оказывается следующим: RI> RВr> RСI> RF. Иное положение наблюдается для является не переходным состоянием, а промежуточным соединением. Величина положительного заряда у реакционного центра зависит не только от количества, расположения и природы других заместителей в ядре, но и от природы замещаемого галогена. Поэтому в активированных ароматических системах атомы галогена могут быть замещены с возрастающей легкостью в ряду I < Вr < СI < F.[13]
Реакционная способность реагента по отношению к галогенпроизводным в реакциях может быть определена как его нуклеофильность. Нуклеофильность агентов зависит от их основности, поляризуемости и сольватации. При переходе от протонных к апротонным растворителям, а также к реакциям в газовой фазе относительная реакционная способность нуклеофилов существенно меняется.
Влияние растворителя в реакциях нуклеофильного замещения очень велико. Переходное состояние SN
1 процесса значительно более полярно, чем исходные вещества. Поэтому увеличение полярности должно приводить к росту скорости диссоциации, а следовательно, и к увеличению скорости процесса в целом. Наряду с неспецифической сольватацией большую роль играет специфическая сольватация, и в первую очередь стабилизация уходящего галогенид - аниона за счет образования водородных связей с растворителем.
Что касается выбора растворителя для SN
2 реакции, то в этом случае необходимо рассмотреть распределение зарядов в исходном и переходном. В соответствии с теорией Хьюза и Ингольда, реакции биомолекулярного замещения можно разбить на четыре типа по способу распределения зарядов и предсказать эффект увеличения полярности среды:
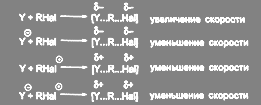 (15) (15)
Наиболее распространенными являются первые два типа реакции. Изложенный подход, однако, не учитывает важности специфической сольватации реагентов, тогда как уменьшение специфической сольватации нуклеофила является одной из основных причин ускорения реакций бимолекулярного замещения типа анион — молекула в апротонных растворителях. Влияние природы растворителя в реакциях нуклеофильного замещения настолько велико, что в ряде случаев определяет протекание реакции по SN
1 или по SN
2 механизму.
К числу полярных растворителей, способствующих протеканию реакций по SN
1 механизму, относятся протонные растворители: вода, спирты, карбоновые кислоты, аммиак. В реакциях нуклеофильного замещения они сольватировать как катионы, так и анионы. Тенденция к образованию водородных связей растет с увеличением кислотности растворителя.
Многие реакции, протекающие в слабо сольватирующих растворителях по бимолекулярному механизму, могут при использовании в качестве растворителя муравьиной или трифторуксусной кислоты идти по SN
1 типу.
К числу нуклеофильных растворителей, которые сольватируют главным образом катион, можно отнести такие апротонные соединения, как ацетон, ацетонитрил, нитрометан, диметилформамид, диметилсульфоксид, диглим и др. Они не сольватируют уходящих галогенид—ионов, а потому не способствуют протеканию SN
1 реакции SN
2 реакции, напротив, легко протекают в этих растворителях, так как в лимитирующей скорость стадии анионов не образуется.
Способностью стабилизации анионов (за счет комплексообразования) обладают кислоты Льюиса (галогениды бора, алюминия, цинка, сурьмы, ртути, серебра, а также ион серебра). Эти соединения применяются обычно как катализаторы для SN
1 реакций. Стабилизация катиона при этом осуществляется путем взаимодействия с реагентом или растворителем.
Помимо уже перечисленных факторов при выборе растворителей необходимо учитывать их растворяющую способность по отношению к реагенту и субстрату. Во многих случаях при осуществлении реакций нуклеофильного замещения в качестве реагентов используются неорганические и органические соли, хорошо растворимые в воде и плохо растворимые в органических растворителях. для проведения таких реакций в гомогенных условиях традиционно применяют растворители, которые проявляют одновременно липофильные и гидрофильные свойства, например метанол, ацетон, этанол, ацетон, диоксан. Трудность при этом заключается в том, что соли менее растворимы в этих растворителях, чем в воде, а органические субстраты обычно менее растворимых в них, чем в углеводородах. Указанную проблему можно частично решить, используя смеси упомянутых выше растворителей с водой. Более эффективным оказывается применение таких диполярных, апротонных, катионсольватирующих растворителей, как диметилсульфоксид, диметилформамид, ацетонитрил, которые хорошо растворяют как соли, так и органические субстраты. Важным методом интенсификации процессов нуклеофильного замещения является межфазный катализ (МФК). Суть метода заключается в искусственном создании двухфазной системы, в которой неполярные и ионные реагенты находятся в разных фазах. Обычно это органическая фаза и водная фаза. Для переноса реагентов (нуклеофилов) служат межфазные катализаторы — источники липофильных катионов. Их роль заключается в образовании липофильных ионных пар "катион катализатора — реагирующий анион, способных к миграции внутрь органической фазы, где и происходит реакция. В применяемых для этой цели апротонных неполярных растворителях, не смешивающихся с водой, реагирующие анионы практически не сольватированы и обладают высокой реакционной способностью. Иногда в качестве органической фазы используют субстрат.[8]
1.3 Замена атома галогена на - ОН, - ОR, - OН, - SН и – SRгруппы
Вода представляет собой слабонуклеофильный реагент. Поэтому галогеналканы в большинстве случаев очень медленно гидролизуются водой до алканов. При обработке алкилгалогенидов кипящей водой образуется смесь алкилгалогенида и алканола:
 (16) (16)
Равновесие может быть сдвинуто вправо под действием гидроксида серебра (суспензия оксида серебра в воде) или гидроксидов щелочных металлов:
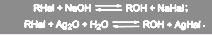 (17) (17)
При этом равновесие не только сдвигается вправо, но реакция значительно ускоряется вследствие высокой нуклеофильности гидроксил-иона. Реакцию часто проводят в спирто-водной или спирто-ацетоновой среде для гомогенизации реакционной массы, так как алкилгалогениды нерастворимы в воде. В зависимости от реагентов и условий проведения реакция может протекать как по SN
1, так и по SN
2 механизму. Побочными реакциями могут быть образование простого эфира за счет взаимодействия галогеналкила со спиртом в щелочной среде и элиминирование с образованием алкена.
В синтетических целях эта реакция используется редко, так как сами обычно получают из спиртов.
Дигалогенпроизводные, у которых оба атома галогена связаны с одним углерода, образуют альдегиды или кетоны. Таким образом получают, например, бензофенон:
 (18) (18)
Для получения альдегидов нельзя применять сильные основания, так как продукт может вступать в альдольную конденсацию.
Соединения, содержащие три атома галогена у одного атома углерода, образуют кислоты:
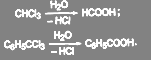 (19) (19)
Практическое применение метода ограничено тем, что тригалогениды труднодоступны.
При проведении гидролиза в присутствии спирта можно сразу получить эфир.
Хлороформ при обработке основанием гидролизуется быстрее, чем дихлорметан или тетрахлорметан и образует не только муравьиную кислоту, но и монооксид углерода.Считают, что реакция протекает через стадию образования дихлоркарбена:
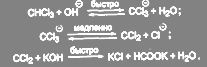 (20) (20)
Дихлоркарбен является очень реакционноспособным соединением. Его промежуточным образованием объясняется целый ряд реакций. Так, например, пятичленные гетероциклы (индол, фуран, тиофен, а также их производные) присоединяют дихлоркарбен с последующей перегруппировкой и ароматизацией полученного продукта, превращаясь в соответствующие 3-хлорзамещенные шестичленные гетероциклические соединения:

Аналогичные реакции присоединения дихлоркарбена известны в ряду сопряженных олефинов, например у циклопентадиена:
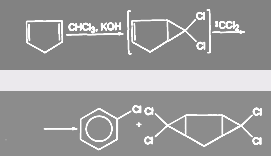
При гидролизе ССl4
образуется фосген:
 (24) (24)
При омылении дигалогенидов не следует применять гидроксиды щелочных металлов, так как образующиеся карбонильные соединения чувствительны к сильнощелочной среде. Омыление проводят в присутствии карбоната кальция или калия, ацетата натрия, формиата или оксалата.
Бензилиденхлориды и бензилиденбромиды гладко гидролизуются до бензалдегидов под действием концентрированной серной слоты. Электронодонорные группы в ядре облегчают гидролиз, электроноакцепторные — затрудняют. В последнем случае следует повысить температуру акции до 120—130 °С (но не выше, т. к. при более высокой температуре альдегиды будут интенсивно окисляться серной кислотой).
Галогениды с повышенной реакционной способностью гидролизуются очень легко. Так, аллилхлорид и бензилхлорид превращаются в аллиловый и бензиловый спирт при кипячении в избытке воды. В зависимости от структуры субстрата и условий проведения реакции могут протекать вторичные процессы. Так, в синтезе мезатона при щелочном гидролизе м-нитрофенилбромметилкарбинола образуется окись м-нитростирола:
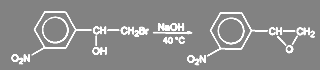 (25) (25)
Аналогичная реакция имеет место в синтезе левомицетина:
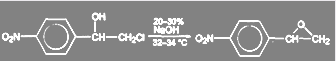 (26) (26)
При образовании п-нитро-α-метоксистирола в спиртовой щелочи проходит реакция элиминирования:
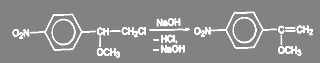 (27) (27)
При получении оригинального отечественного противоопухолевого препарата допан замену галогена в гетероароматическом ядре проводят при кипячении с соляной кислотой. Следует обратить внимание на то, что хлор в хлорэтильных группах при этом не обменивается. Выход очищенного продукта 85—86%.
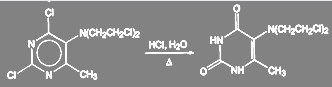 (28) (28)
Значительно чаще в синтезе лекарственных препаратов используется нуклеофильная замена атома галогена на ОАIk-группу. Реакцию проводят либо со спиртом в щелочной среде, либо с алкоголятом.
Реакция обычно требует активированных субстратов, в противном случае выход целевого продукта (эфира) может оказаться низким за счет побочных процессов. В случае фенольных нуклеофилов (АгОН) реакция катализируется солями меди, в присутствии которых нет необходимости в наличии активирующих групп. Считают, что в этом случае реакция идет за счет активных реагентов АгОСu. Выходы эфиров по этому методу обычно высокие. Более того, в качестве нуклеофилов иногда используют соли кислот RСООН. При обработке арилгалогенидов бензоатом меди (I) в диглиме или ксилоле при температуре от 140 до 160 °С с высокими выходами получают арилбензоаты. При получении сульфапиридазина 3-хлор-6-сульфаниламидопиридазин нагревают 9 ч при 130 ºС и давлении 0,7—0,8 МПа с большим избытком едкого кали в среде метанола:
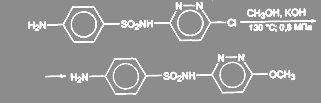 (29) (29)
Аналогично идет процесс в синтезе сульфалена:
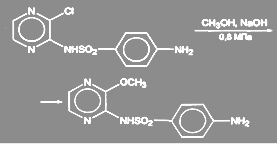 (30) (30)
В синтезе сульфадиметоксина замена хлора в 4-амино-2,6-дихлорпиримвдине происходит при 20-часовом кипячении в метанольном растворе NаОН:
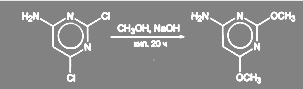 (31) (31)
В ряду пятичленных гетероароматических соединений такие реакции проводят лишь при активации галогена электроноакцепторными заместителями:
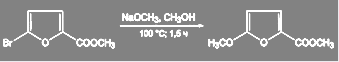 (32) (32)
Реакция получения простых эфиров по Вильямсону является одним из примеров успешного применения метода межфазного катализа для синтеза органических соединений. В условиях МФК она дает более высокие выходы продуктов, протекает с большей скоростью и является более простой в техническом исполнении. В большинстве случаев при проведении процессов традиционным методом необходимо предварительно получить алкоголят, что требует использования таких сильных оснований, как металлический натрий, амид натрия, а также предварительного обезвоживания реагентов и растворителей.[5]
В двухфазном синтезе простых эфиров в качестве основания используется концентрированный водный раствор едкого натра (обычно 50%). Спирт в растворе депротонируется по действием гидроксил-аниона вводной фазе или на границе раздела фаз. Растворение аниона алкоголята в органической фазе происходит за счет образования ионной пары с липофильным катионом межфазного катализатора (чаще всего четвертичной соли аммония). Тот факт, что в органическую фазу переходит алкоголят тетраалкиламмония, а не его гидроксид, объясняется тем, что ОН-
более эффективно сольватируется водой, чем алкоголят-анион и предпочтительно остается в водной фазе.
Наблюдаемое повышение скорости образования простого эфира объясняется несколькими факторами. Главная компонента энергетического барьера в реакциях SN
2 — удаление молекул растворителя от нуклеофила. В традиционном способе (в гомогенных условиях) десольватация аниона нуклеофила затруднена, особенно в протонньихрастворителях. В условиях МФК анион в виде ионной пары с большим липофильным катионом аммонийной соли катализатора в неполярной органической фазе сольватирован мало. Вторым важным преимуществом МФК является то, что реакционная способность алкоголят-аниона, связанного с большим по размерам катионом МФ-катализатора, оказывается выше, чем у аналогичного иона в алкоголяте калия или натрия.[7]
Приведем несколько примеров успешных опытов получения простых эфиров в условиях МФК:
 (33) (33)
Известно несколько примеров аналогичных подходов в синтезах диариловых эфиров. Так, введение в качестве катализатора хлорида гексадецилтриметиламмония (ГДТМАХ) в реакцию п-нитрохлорбензола с п-метоксифенолом в присутствии 25% раствора КОН повышает выход эфира с 67% до 98%:
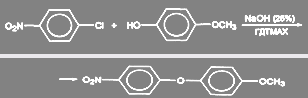
Если вместо гидроксил- или алкоголят-аниона взять гидросульфид-меркаптяд-ионы, то образуются соответствующие арилтиолы и тиоэфиры.
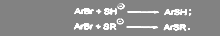 (36) (36)
Активированные арилгалогениды обычно дают хорошие выходы, но в ряде случаев побочные реакции являются конкурентными. Под действием SАг-
можно получить диарилсульфиды. В реакцию с SАг-
вступают даже неактивированные арил- и алкилгалогениды, если проводить ее в полярных апротонных растворителях (диметилформамид, диметилсульфоксид) Диарилсульфиды получаются с высокими выходами при обработке неактивированных арилиодидов SАг-
в жидком аммиаке. Реакции с галогеналкилами обычно проводятся в среде этанола:
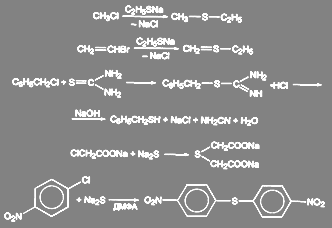 (37) (37)
Симметричные и смешанные сульфиды могут быть получены и в условиях МФК. Так, например, с высокими выходами образуются несимметричные тиоэфиры из первичных и вторичных алкилбромидов при их взаимодействии с меркаптанами и тиофенолами в присутствии водных растворов щелочи. В качестве катализаторов межфазного переноса использовались ониевые соли, краун-эфиры и криптаты. детально изученный механизм реакции тиофенола с н-бромоктаном в щелочной двухфазной системе подтверждает SN
2 характер замещения:
 (38) (38)
1.4 Замена атома галогена на группы -NН2
, -NНR, -NR2
Алкил- и арилгалогениды могут взаимодействовать с аммиаком и аминами. Эти реакции можно рассматривать также и как частный случай N-алкилирования.
Если нагревать алкилгалогенид со спиртовым (реже водным или водноспиртовым) раствором аммиака, первичного или вторичного амина под давлением и при высокой температуре (в автоклаве), то образуется смесь солеи соответствуюших аминов (первичных, вторичных, третичных), а так же четвертичных солей аммония (реакция Гофмана):
 (39) (39)
Вместо аммиака можно использовать первичные, вторичные или третичньие амины (реакция Меншучкина) и получать смешанные аминосоединения:
 (40) (40)
Третичные алкилгалогениды в этих реакциях обычно не применяют, так как в условиях реакции идет элиминирование с образованием алкенов.
Выход первичного амина можно повысить, применяя большой избыток аммиака и добавляя карбонат или хлорид аммония. Однако даже в этом случае образуется смесь соединений, которые приходится разделять.
Поскольку для синтеза биологически активных веществ особенно важно иметь индивидуальные и чистые вещества строго определённой структуры, особое значение приобретают те методы и приёмы, которые позволяют достичь такого результата.
Один из возможных путей синтеза вторичных аминов без примеси третичных ясен из схемы:
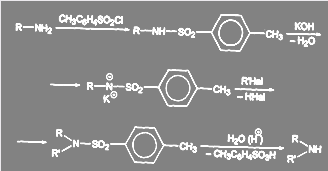 (41) (41)
Другим способом получения вторичных аминов является синтез через азометины:
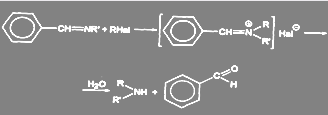 (42) (42)
Первичные амины можно получить гидролизом и гидразинолизом алкилфталимида (синтез Габриэля):
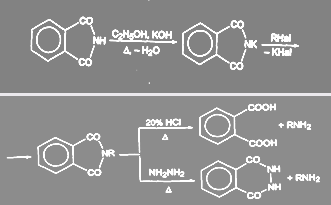
Преимущество гидразинолиза перед гидролизом состоит в том, что гидролиз приходится проводить при высоких температурах под давлением, в то время как взаимодействие с гидразином идет при нормальном давлении.
Допустимые выходы первичных аминов дают лишь α-галогенкарбоновые кислоты при действии большого избытка аммиака. Таким образом получают:
 (45) (45)
Реакцию ведут, нагревая при 40—50 °С соответствуюшую галогензамещенную кислоту с концентрированным водным раствором аммиака и карбоната аммония. Выходы α-аминокислот — 60—70%.
В неактивированных галогенпроизводных ароматического ряда галоген может быть замещен на аминогруцпу действием раствора аммиака при высокой температуре и давлении в присутствии катализатора (Сu2
O, СuSO4
, и т.д.). Другой способ состоит в действии амид-иона в жидком аммиаке. Реакция в этом случае идёт через образование дегидробензола.[2]
Условия проведения реакции в активированных соединениях зависят от степени активации галогена:
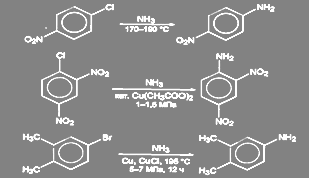 (46) (46)
3-хлор-6-(п-аминбензолсульфаниламидо)-пиридазин получают из 3,6-дихлорпиридазина. Реакцию ведут в среде циклогексанола с добавкой пиридина и значительного количества поташа (2 моля на 1 моль) при температуре 135 – 160 ºС: T5r
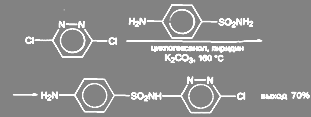 (47) (47)
Получение 2-метил-4-(α-метил-δ-диэтиламинобутиламино)-6-метоксихинолина в производстве трихомонацида ведут в более жёстких условиях, в этом случае требуется двойной избыток диэтиламино-4-аминопентана и температура 185 – 190 ºС:
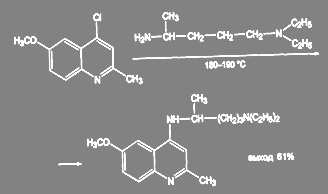 (48) (48)
При получении антидеприсанта азафена аминирование ведут дигидрохлоридом N-метилпиперазина:
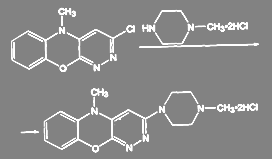 (49) (49)
Получение амидов из хлорангедридов кислот идёт в мягких условиях. Так, получение диакарба проводят при температуре 3-4 ºС действием охлаждённого раствора аммиака на соответствующий сульфохлорид:
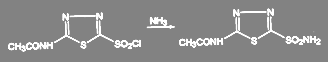 (50) (50)
Аналогичные реакции имеют место при получении метилуретана:
 (51) (51)
1.5 Замена атома галогена на - СN и - SO3
Na
Замена галогена на СN-группу является относительно простым способом удлинения углеродной цепи. Этот способ, однако, неприменим для пространственно затрудненных субстратов. Первичные галогениды, а также бензил- и аллилгалогениды дают хорошие выходы нитрилов. В случае вторичных алкилгалогенидов выходы средние. С третичными галогенидами реакция не идет, так как в этих условиях проходит реакция не замещения, а элиминирования, Для проведения синтеза пригодны многие растворители, но наилучшие результаты достигаются в диметилсульфоксиде (ДМСО).[6]
Реакционная способность алкилгалогенидов увеличивается в ряду CI< Вr <I, что позволяет проводить реакцию избирательно:
 (52) (52)
Поскольку цианид-ион является амбидентнsм ионом, реакция может идти по двум направлениям — с образованием нитрилов и изонитрилов:
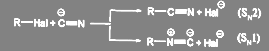 (53) (53)
В случае первичных алифатических галогенидов и бензилгалогенидов в спиртах и водно-спиртовых смесях нежелательная примесь изонитрилов не образуется или образуется в очень малых количествах (легко обнаруживается по крайне неприятному запаху).
В случае ароматических галогензамещенных реакцию следует вести в апротонных растворителях. Галогенбезолы могут также превращаться в соответствующие фенилцианиды при нагревании до 200 °С с цианидом меди (1) в растворе пиридина:
 (54) (54)
Реакционноспособные алкилгалогениды переводят в нитрилы кипячением галогенида и цианида натрия в сухом ацетоне с добавлением небольшого количества (0,05 моля на 1 моль субстрата) иодида натрия. Для инетных галогенидов в качестве растворителя используют 70—90% спирт или триэтиленгликоль. Правильный выбор растворителя во многом определяет успех реакции. Так, например, алифатические нитрилы можно получать с высоким выходом в ДМСО или ДМФА. Наибольшие примеси изонитрила можно гидролизовать в кислой среде и таким образом отделить от основного продукта, так как нитрилы гидролизуются в значительно более жестких условиях.[17]
Нитрилы являются важными промежуточными продуктами в синтезе многих химико-фармацевтических препаратов, так как легко превращаются в амиды карбоновых кислот, карбоновые кислоты, амины:
 (55) (55)
Как сам бензилцианид, так и получающиеся из него фенилуксусная кислота и -фенилэтиламин широко используются в синтезе биологически активных соединений.
Из монохлоруксусной кислоты можно получить циануксусную кислоту (для синтеза теобромина, теофиллина, кофеина и др.) и используемую во многих синтезах малоновую кислоту:
 (56) (56)
Замещенные бензилцианиды также широко используются для синтеза лекарственных веществ:
 (57) (57)
При синтезе папаверина З,4-диметоксифенилацетонитрил получают так называемым цианметилированием вератрола. Реакция, вероятно, идет как минимум в две стадии: на первой стадии образуется 3,4-диметоксибензилхлорид, который под действием цианида натрия превращается в нитрил:
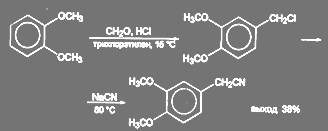 (58) (58)
В ряду пятичленных гетероароматических соединений реакция идет в довольно жестких условиях:
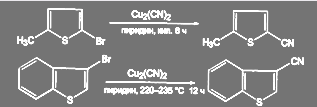 (59) (59)
Галоген в пиридиновом ядре достаточно подвижен, что может быть использовано, в частности, при синтезе никотиновой кислоты:
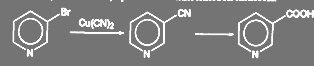 (60) (60)
Замещение галогена на цианид-ион — это одна из первых реакций, в которых нашел применение межфазный катализ.
Установлено, что реакция нуклеофильного замещения происходит в органической фазе и эта бимолекулярная стадия определяет скорость всего процесса. Реакция идет быстрее с первичными, чем со вторичными алкилгалогенидами. В случае вторичных алкилгалогенидов с реакцией замещения конкурирует процесс элиминирования.[18]
Реакция с цианид-ионом в условиях МФК идет часто даже лучше, чем в диполярных апротонных растворителях. Например, из втор-хлороктана в условиях МФК образуется 85—90% продуктов замещения и 10—15% продуктов элиминирования, в то время как в гомогенной среде в диметилсульфоксиде образуется только 70% втор-цианооктана. Помимо четвертичных ониевых солей при цианировании в качестве катализаторов успешно применены краун-эфиры и криптаты.
Преимущества проведения реакции цианирования в условиях МФК хорошо иллюстрируются на примере производства цианистого бензила, важного полупродукта в синтезе целого ряда лекарственных препаратов.
 (61) (61)
По традиционной технологии указанную реакцию проводили в 75% водном этаноле при 76—78 °С. Выход цианистого бензила составлял 77— 79% (в расчете на хлористый бензил). Помимо основного продукта в реакпии образовывались примеси бензиламина, бензилового спирта, бензилового эфира, фенилуксусной кислоты и других веществ, что затрудняло отделение и очистку целевого продукта.[17]
Проведение процесса цианирования хлористого бензила в двухфазной системе С6
Н5
СН2
СI — водный раствор NаСN позволило увеличить выход продукта реакции до 95—96%. В качестве катализаторов процесса в настоящее время используют бензилтриэтиламмоний хлорид или бензилдиметилформвламмоний хлорид, которые образуются непосредственно в реакционной массе, при добавлении триэтиламина или диметилформамида (около 0,5% мольн.):
 (62) (62)
В двухфазной системе в мягких условиях проведено цианирование ряда арилгалогенидов. Однако для этих соединений наряду с межфазными катализаторами (четвертичными аммониевыми солями и краун-эфирами) требуется вводить сокатализаторы. В качестве последних используются комплексы никеля и солей палладия с арил- или алкилфосфинами.
Важной реакцией, использующейся при синтезе лекарственных веществ, является реакция между сульфитом щелочного металла и органическим галогенидом (реакция Штреккера):
 (63) (63)
Выходы сульфокислот составляют 70-90 % при использовании первичных алкилгалогенидов 20-25% для вторичных. Третичные алкилгалогениды превращаются в олефины. В реакцию вступают также галогензамещённые кислоты, спирты, кетоны и ароматические соединения с подвижным галогеном:
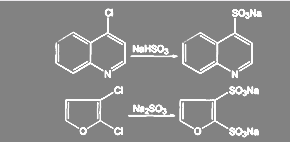 (64) (64)
2. Нуклеиновое замещение сульфогруппы
2.1 Общие сведения о процессе
Нуклеиновое вытеснение сульфогруппы является важнейшим способом синтеза гидрокси- и аминосоединений в ароматическом ряду. Особенно часто эта реакция используется для промышленного получения гидрокси соединений. Поскольку процесс обычно поводят при взаимодействии со щёлочью при высокой температуре, то он известен так же известен под названием щелочного плавления.
Реакция идёт в соответствии со схемой:
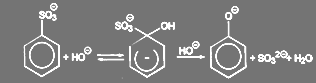 (65) (65)
В зависимости от соотношения констант скоростей первой и второй стадии процесса реакция идет по второму (первый по гидроксилу) или третьему (второй по гидроксилу) порядку. Энергия активации велика и составляет 145—200 ж/моль при 300—340 ºС.[20]
Обычно для щелочного плавления применяют растворы сульфокислот или их пасты, получающиеся при выделении солей сульфокислот. В качестве щелочи обычно используют едкий натр. Едкое кали как более дорогое сырье используют лишь в тех случаях, когда едкий натр оказывается недостаточно реакционноспособным. В некоторых случаях используют смесь едкого натра и едкого кали. Такая смесь имеет более низкую температуру плавления, чем каждая из взятых щелочей. Взаимодействие протекает по уравнению:
 (66) (66)
Наличие в технической щелочи значительных примесей минеральных солей приводит к образованию в плаве комков, затрудняющих перемешивание и значительно ухудшающих качество продукта вследствие местных перегревов реакционной массы. Содержание в щелочи минеральных солей поэтому не должно превышать 10%. Примесь хлората натрия в щелочи недопустима, так как она может вызывать возгорание плава.[4]
Щелочное плавление в промышленности осуществляется тремя способами:
1 Открытое щелочное плавление, т. е. сплавление пасты натриевой или калиевой соли сульфокислоты со щелочью или ее концентрированным раствором при атмосферном давлении;
2 Автоклавное щелочное плавление проводится под давлением в автоклавах. В этом случае используют водные растворы солей сульфокислот. Метод применяется в тех случаях, когда нужно заместить гидроксилом лишь одну из нескольких сульфогрупп, имеющихся в молекуле исходного соединений. Этот метод используют также при щелочном плавлении аминосульфокислот, так как при проведении этого процесса открытым способом аминогруппа также замещается гидроксигруппой. Практически на каждый моль сульфосоли загружают не 2, а от 2,1 до 3,5 молей щелочи;
3 Автоклавное щелочное плавление с известью. Этот способ используется главным образом при щелочном плавлении сульфокислот антрахинона и в других аналогичных случаях, когда обычные методы приводят к введению в ароматическое ядро двух орто-расположенных гидроксигрупп.[4]
Реакцию проводят в присутствии КNО3
(так называемый окислительный плав) и 200 °С.
 (67) (67)
Важнейшими параметрами, определяющими результат щелочного плавления, являются температура ведения реакции и концентрация щелочи.
Едкий натр — не только реагент, но и среда, в которой протекает превращение. Поэтому во многих случаях используется значительный избыток щелочи. Едкий натр плавится при температуре 327,5 ºС. Чтобы обеспечить подвижность плава при более низких температурах, используют концентрационные растворы щелочи (40—42% раствор или 70—73% раствор, упаренный до 80—85%) или же добавляют в щелочь небольшое количество воды. Так, едкий натр, содержащий 10% воды, плавится при температуре 270—290 °С. В процессе плавки вода испаряется и температура реакционной массы поднимается.[13]
Сульфокислоты бензольного ряда обменивают сульфогруппу на гидроксил в жестких условиях, при температуре 300—340 °С. Таким образом получают фенол и резорцин. При синтезе прозерина щелочное плавление натриевой соли м-диме проводят при 275—290 °С.
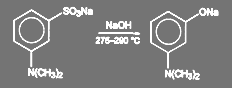 (68) (68)
Активированная сульфогруппа обменивается на гидроксигруппу в более мягких условиях. При наличии в молекуле нескольких сульфогрупп можно подобрать температуру процесса и концентрацию щелочи таким образом, что замещаться будет только наиболее активированная сульфогруппа. Электронодонорные заместители в о- и п-положениях к сульфогруппе будут затруднять реакцию щелочного плавленяя, а электроноакцепторные — облегчать.[10]
Нитросульфокислоты и хлорсульфокислоты ароматического ряда в реакциях щелочного плавления не используют вследствие образования значительного количества побочных продуктов.
Обработка готового щелочного плана включает нейтрализацию избыточной щелочи, отделение сульфита, выделение и очистку гидроксисоединений.
Готовый щелочной плав разбавляют водой. Эту операцию называют гашением щелочного плава. Для выделения сульфита натрия в твердом со стоянии полученную массу нагревают до 80—90 °С, а затем отфильтровывают и промывают на фильтре выпавший сульфит. Гидроксисоединение выделяют при подкислении щелочных маточников. Таким методом пользуются, например, при получении фенола и 2-нафтола.
В тех случаях, когда сульфит отделяют в виде раствора, плав разбавляют большим количеством воды, достаточным для его полного растворения. Избыток щелочи нейтрализуют кислотой. Нейтрализацию ведут до полного выделения гидроксисоединения, которое отделяют от раствора сульфита отстаиванием или фильтрацией.[11]
В некоторых случаях более выгодным оказывается проводить разложение сульфита кислотой:

Для полного удаления сернистого газа из реакционной массы процесс проводят при 80—100 °С и хорошем перемешивании. Выделяющийся сернистый газ поглощают раствором щелочи таким образом, чтобы получить сульфит натрия, который используется в качестве сырья во многих производствах.[12]
Аппаратура в процессах щелочного плавления работает в очень тяжелых условиях. Установлено, что расплавленные щелочи в присугствии органических веществ агрессивно действуют на черные металлы. Металл становится хрупким. По механическим свойствам и коррозионной стойкости в среде концентрированных щелочей при высокой температуре лучшим металлом является никель. Однако из чистого никеля аппаратуру делают редко, так как она получается слишком дорогой. Обычно котлы для щелочного плавления изготавливают из легированного чугуна или легированной стали, содержащей никель, хром и молибден в качестве легирующих присадок. Однако даже при использовании легированных чугунов срок службы плавильных котлов не превышает 2—З лет. Вследствие необходимости поддержания высокой температуры обогрев плавильных котлов обычно ведется топочными газами. В районах с дешевой электроэнергией можно использовать также электрообогрев. Для предотвращения пригорания щелочного плава при подвижных плавах используют пропеллерные мешалки, а при вязких — якорные. Для щелочной плавки под давлением применяются обычные автоклавы.
Гашение плана проводят в стальных котлах с рамными или лопастными мешалками. Аппараты для подкисления разбавленного плана обязательно должны быть защищены от кислотной коррозии — футерованы или освинцованы.[13]
Замещение сульфогруппы аминогруппой протекает при сплавлении натриевой соли сульфокислоты с амидом натрия:
 (70) (70)
Большого практического применения эта реакция не получила.
Замена активированной сульфогруппы может быть проведена при обработке водным раствором аммиака под давлением:
 (71) (71)
Примером может служить промышленное получение аминоантрахинонов из соответствующих сульфокислот:
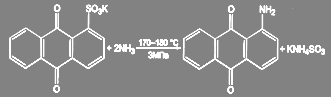 (72) (72)
В тех случаях, когда сульфогруппа обладает особенно высокой нуклеофильной подвижностью, например:
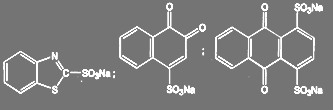 (73) (73)
Взаимодействие с аммиаком проходит при комнатной температуре и атмосферном давлении.
При взаимодействии с гидразидом натрия сульфогруппа в мягких условиях может быть замещена на гидразо-группу:
 (74) (74)
Так же как и атом галогена, сульфогруппа может быть замещена не только гидроксильной или аминогруппой, но и другими нуклеофилами. При нагревании до 200—300 °С щелочные соли аренсульфокислот могут быть превращены в амины, фенолы, тиолы, карбоновые кислоты или нитрилы:
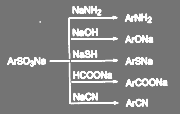 (75) (75)
2.2 Примеры осуществления нуклеофильной замены сульфогруппы в промышленности
Производство фенола. Одним из промышленных методов производства фенола является щелочное плавление натриевой соли бензолсульфокислоты:
 (76) (76)
При выдержке 30 мин и 15% избытке щелочи оптимальная температура этого процесса З2О—ЗЗО °С. В случае применения едкого кали температура щелочного плавления 280—285 °С, а при использовании смеси 80% NаОН и 20% КОН — 300 °С. Выход фенола составляет около 96%.
В реактор загружают 70—75% раствор щелочи и упаривают его до концентрации щелочи 80—85% в плавильных котлах. Из стальных мерников в котел постепенно вводят раствор бензолсульфоната. Реакционная вода, образующаяся в процессе плавления, испаряется с некоторым количеством фенола (1—1,5% общего количества). Пары воды, содержащие фенол, конденсируются в скрубберах, орошаемых водой, и образующийся конденсат передают на выделение фенола. По окончании операции щелочной плав сливают в наполненный водой стальной гаситель, снабженный мешалкой. Пары воды, выделяющиеся из гасителя, содержат часть фенола. Их конденсируют и конденсат присоединяют к фенольным водам.
Выход фенола на стадии плавления — 96% от теоретического. При улавливании фенола из паров, выделяющихся в плавильном котле, выход на стадии плавления можно повысить до 97—98%.
Выход на стадии гашения 99% от теоретического. При улавливании фенола из паров, выделяющихся из гасителя, выход может достигать 99,5—99,8%.[14]
Производство резорцина. Щелочное плавление сухих м-дисульфоната бензола и едкого натра проводят в стальном литом котле емкостью около 1 м3
. Котел снабжен мощной (64 кВт) лопастной мешалкой, укрепленной в подпятнике и вращающейся со скоростью 20 об/мин. Выгрузка плана производится через широкое выгрузочное отверстие в днище котла. Обогрев котла электрический. Особая трудность аппаратурного оформления процесса получения резорцина связана с тем, что в процессе щелочного несколько раз меняется консистенция реакционной массы. Загружаемая в аппарат порошкообразная смесь при 210—220 ºС превращается в вязкую пластичную массу, которая разжижается при температуре выше 220°С; при температуре 290 °С масса снова густеет и снова превращается в порошкообразный твердый продукт, который при 310 ºС превращается в тестообразную массу, а при 340 ºС затвердевает. Эти изменения вязкости реакционной массы обусловлены протеканием процесса в несколько стадий. Вначале замещается одна сульфогруппа и из реакционной массы отгоняется вода, а затем происходит замещение второй сульфогруппы и отгонка образовавшейся воды:

Резорцин, в отличие от фенола, хорошо растворим в воде и в водных растворах солей, а потому не осаждается при подкислении растворенного в воде плава. В связи с этим после отделения сульфита из нейтрализованного раствора плава резорцин извлекают экстракцией органическими растворителями.[15]
3. Особенности техники безопасности при проведении процессов нуклеофильной замены галогена и сульфогруппы
Во многих случаях процессы нуклеофильного замещения галогена (особенно при замене на -ОН, -ОR, -NН2, -NНR-группы) проводят при температурах более высоких, чем температура кипения реагентов или среды. Поэтому в тех случаях, когда реакцию проводят в жидкой фазе, приходится использовать автоклавы или другие аппараты для проведения процессов под давлением.[16]
Установка и эксплуатация автоклавов, а также проектирование и эксплуатация автоклавных отделений должны проводиться в соответствии с требованиями "Правил устройства и безопасной эксплуатации сосудов, работающих под давлением". При этом необходимо установить безопасный режим работы автоклава, в том числе допустимые скорости повышения и снижения температуры и давления.
Аппараты для проведения технологических процессов с применением сильнодействующих и ядовитых веществ (цианиды, метиловый спирт и т. д.) должны размещаться в боксах, укрытиях, камерах, оборудованных местными отсосами и необходимыми устройствами по предупреждению распространения вредностей или в отдельных изолированных помещениях.[17]
К работе с метанолом допускаются лишь лица, прошедшие специальный инструктаж. При работе с метанолом необходимо руководствоваться "Правилами по перевозке, хранению и применению метанола", а также "Правилами безопасности для производства медицинской промышленности". Получение метанола со склада, хранение и расходование его проводятся под строжайшим контролем ответственных лиц, так как метанол является сильным ядом. Запрещается совместное хранение (в одном складе) метанола с этиловым спиртом. Все оборудование, в котором происходят процессы с применением метанола, должно быть герметизировано и снабжено отсосами. Лабораторные работы с применением метанола могут проводиться только в вытяжных шкафах. Для работы в заводских цеховых лабораториях метанол может выписываться в размере суточной потребности, остаток должен сдаваться на склад или храниться в опечатанном шкафу.[18]
Применение метанола допускается лишь в тех случаях, когда он не может быть заменен менее токсичным веществом.
Во многом аналогичные правила установлены для работы с цианистыми соединениями и соединениями мышьяка.
Соответствующие отделения должны иметь поглотительные установки для обезвреживания токсичных выделений, средства для контроля за состоянием воздушной среды, индивидуальные средства защиты и т. д.
В отделениях цианирования не допускается устройство заглубленных аппаратов и приямков. Маточники, сточные воды, воды от промывки аппаратов и смыва полов должны обезвреживаться на локальных установках.
В помещении цианирования должны находиться не менее двух человек — аппаратчик и мастер — для наблюдения за процессом и оказания необходимой помощи в случае аварии.
Во всех случаях применения токсичных соединений или соединений, образующих взрыво-пожароопасные смеси с воздухом должна строго контролироваться герметичность оборудования. Необходимо помнить, что амины являются нервными и кровяными ядами, спирты обладают наркотическим действием, щелочь при попадании на кожу (особенно на слизистую оболочку) вызывает химический ожог, вдыхание пыли Nа2
SО3
и других солей вызывает раздражение дыхательных путей и т. п.[19]
Аммиак, спирты и многие растворители, используемые в процессах замещения галогенов, образуют взрыво- и пожароопасные смеси с воздухом.
Многие фенолы и амины являются сильными нервными и кровяными ядами.
Заключение
нуклеофильный галоген сульфогруппа синтез
Материал, рассмотренный в данной работе должен помочь будущему химику – технологу освоить методы тонкого органического синтеза, правильно выбрать оптимальную химическую схему синтеза целевого продукта. Данная работа посвящена главным образом рассмотрению нуклеофильной замены галогена и сульфогруппы на другие функциональные группы. Реакции нуклеофильного замещения галогена являются весьма важными и распространенными в органическом синтезе и синтезе лекарственных веществ, а так же витаминов. Они используются в синтезе димедрола, фенацетина, азалина, сульфапиридазина, сульфалена, сульфадиметоксина, диакарба, азафена, папаверина, левомицетина, витаминов В2
и В6
. Так же этот метод имеет большое значение в синтезе пептидов, антибиотиков, модификаций сахаров. [20]
Список использованных источников
1 Белобородов В.Л., Зурабян С.Э., Лузин А.П., Тюкавкина Н.А. Органическая химия. – М.: Дрофа, 2002. – Кн.1 : Основной курс. – 640 с.: ил.
2 Беляев Е.Ю. Практическая органическая химия. — Изд-во Красноярского университета, 1996. — 439с.
3 Березин Б.Д., Березин д.Б. Курс современной органической химии. —Химия, 1999. — 768 с.
4 Джальберт Э. Сульфирование органических соединений. — М.: Химия,—414 с.
5 Ингольд К Теоретические основы органической химии. — М.: Мир,
1973. — 1055 с.
6 Карапетьянц М.Х., Дракин С. И. Общая и неорганическая химия. —
4-е изд. — М.: Химия, 2000. — 592 с.
7 Липович В.Г., Полубенцева М. Ф. Алкилирование ароматических углеводородов. — М.: Химия, 1985. — 272 с.
8 Лисицын В.Н. Химия и технология промежугочных продуктов. — М.:Химия, 1987. — 368 с.
9 Марч Дж. Органическая химия, углубленный курс для университетов: В 4 т. — М.: Мир, 1987—1988.
10 Матьеж, Панико Р., Вейль-Рейналь Р. Изменение и введение функций в органическом синтезе. — М.: Мир, 1980. — 488 с.
11 Машковский МЛ Лекарственные средства: В 2 т. — 14-е изд. перераб. и доп. — М.: Медицина, 2000. Т 1. 540 с.; Т 2. 608 с.
12 Муганлинский Ф.Ф., Трегер Ю.А., Люшин М.М. Химия и технология галогенорганических соединений. — М.: Химия, 1991. — 272 с.
13 Пассет Б.В. Основные процессы химического синтеза биологически активных веществ (БАВ): Учебник. – М.: ГЭОТАР – МЕД, 2002. – 376с.
14 Черных В.П., Гридасов В.И., Гриценко И.С., Колесникова Т.А. Руководство к лабораторным и семинерским занятиям по органической химии. – Х.: Выш. шк.,изд – во при ХГУ, 1989, - 348с.: схем.
15 Яхонтов Л.Н., Глушков Р.Г. Синтетические лекарственные средства. —М.: Медицина, 1983. — 272 с.
16 http://www.krugosvet.ru/enc/nauka_i_tehnika/himikal
17 www.muctr.ru.newhtml/041-335.htm
18 www.rihophe.ru/lab61/staff.html
19 www.niopik.ru/about/scientists/Vorozhtsovz
20 www.spepa.ru/learnimgzao/z9.html
|