| Глава 1. Литературный обзор
1.1 Экологические характеристики гетерогенных катализаторов
На сегодняшний день каталитические процессы составляют основную массу деструктивных процессов переработки нефти. Их популярность, помимо многих других факторов, обусловлена снижением экологической нагрузки при проведении каталитических процессов переработки углеводородных систем по сравнению с термическими процессами. Необходимость увеличения глубины переработки нефти в России с целью увеличения выхода светлых фракций, и в частности, автомобильных бензинов, обусловливает создание новых мощностей по производству моторных топлив.
В Западной Европе ужесточение экологических требований к качеству потребляемых автопромом бензинов произошло около 15 лет назад. В России переход на международные стандарты пытаются осуществить в настоящее время, однако наблюдается острая нехватка в качественных высокооктановых моторных топливах. Сроки введения экологических нормативов на выбросы автомобилями приведены в таблице 1 [1].
Таблица 1- Сроки введения экологических нормативов на выбросы автомобилями и требований к качеству бензинов
| Норма
|
Легковые автомобили
|
Автомобильный бензин
|
| |
Западная Европа
|
Россия
|
Западная Европа
|
Россия: требования
|
| |
|
|
|
Тех.регламента
|
Нормативной документации
|
| Евро-2
|
1996
|
22 апреля 2006г.
|
1994
EN 228:1993
|
До 31.12 2008г.
|
1997г.
ГОСТ Р 51105
|
| Евро-3
|
2000
|
1 января 2008г.
|
2000
ЕN 228:1999
|
1 января 2009г.
|
2002 г.
ГОСТ Р 51866
|
| Евро-4
|
2005
|
1 января 2010г.
|
2005
ЕN 228:2004
|
1 января 2010г.
|
2005 г.
ТУ 38.401-58-350-2005
|
| Евро-5
|
2009
|
1 января 2014г.
|
2009
ЕN 228:2004
|
1 января 2013г.
|
2007г.
Изм.1 к ГОСТ Р 51866
|
Изменение № к Тех. Регламенту: Евро-2 до 31 декабря 2009 г.; Евро-3 с 1 января 2010 г.; Евро-4 с 1 января 2012 г.; Евро-5 с 1 января 2015 г.
Основные требования к качеству автомобильных бензинов, выпускаемых в оборот и находящихся в обороте, на период до 2015 года определены специальным техническим регламентом (ТР) [2], принятым Правительством РФ 27 февраля 2008 г. (табл.1). В сентябре указанный регламент должен был вступить в силу, но этого не произошло. В таблице 2 представлены требования технического регламента к качеству автомобильных бензинов.
Таблица 2 - Требования к характеристикам автомобильного бензина
| Характеристики
автомобильного бензина
|
Нормы в отношении
|
| |
класса 2
|
класса 3
|
класса 4
|
класса 5
|
| Массовая доля серы, мг/кг, не более
|
500
|
150
|
50
|
10
|
| Объемная доля бензола, %, не более
|
5
|
1
|
1
|
1
|
| Концентрация железа, мг/дм3
, не более
|
отсутствие
|
отсутствие
|
отсутствие
|
отсутствие
|
| Концентрация марганца, мг/дм3
, не более
|
отсутствие
|
отсутствие
|
отсутствие
|
отсутствие
|
| Концентрация свинца, мг/дм3
, не более
|
отсутствие
|
отсутствие
|
отсутствие
|
отсутствие
|
| Массовая доля кислорода, %, не более
|
-
|
2,7
|
2,7
|
2,7
|
| Объемная доля углеводородов, %, не более:
|
|
|
|
|
| ароматических
|
-
|
42
|
35
|
35
|
| олефиновых
|
-
|
18
|
18
|
18
|
| Октановое число:
|
|
|
|
|
| по исследовательскому методу, не менее
|
92
|
95
|
95
|
95
|
| по моторному методу, не менее
|
83
|
85
|
85
|
85
|
Требования достаточно жесткие, и обеспечить их соответствие на сегодняшний день могут лишь не многие нефтеперерабатывающие предприятия России [3]. Только использование процессов алкилирования и изомеризации для производства бензинов может улучшить их качество до требуемых нормативов. Поэтому строительство этих установок в составе Российских НПЗ является гарантом получения моторного топлива, соответствующего международным стандартом.
Одной из причин, по которой Российские нефтеперерабатывающие предприятия не спешат строить новые установки для получения качественного высокооктанового топлива, являются неизбежные первоначальные капитальные затраты на закупку технологического оборудования. Зарубежные технологии дороги, отечественные не многочисленны и не всегда безупречны. В этих технологиях одним из ключевых звеньев, влияющим как на технологические параметры процесса (температуру, давление, расход реагентов, и как следствие - на капитальные затраты), так и на экологические показатели процесса (состав получаемых продуктов и количество токсичных выбросов), является грамотный подбор катализаторов.
Современные катализаторы глубокой переработки нефти должны отвечать следующим основным требованиям: активностью и селективностью по отношению к проводимому процессу, достаточной механической прочностью, доступностью, приемлемой стоимостью, большим сроком службы. Именно срок службы катализаторов зачастую определяет, будет ли катализатор внедрен в промышленное использование, или сможет эксплуатироваться лишь в масштабах лаборатории.
Не на последнем месте среди критериев отбора промышленных катализаторов находятся такие их свойства, как способность к регенерации и параметры этого процесса. Ведь сложности в осуществлении регенерации, повышенные энергетические затраты, большая продолжительность, наличие токсичных веществ в составе газов, отходящих из регенератора – все эти факторы влияют не только на экономические затраты, но и, прежде всего, на экологические показатели процесса.
1.2 Причины потери активности цеолитсодержащих катализаторов
в ходе эксплуатации
В настоящее время, подавляющее большинство термокаталитических процессов нефтепереработки (около 90 %) основано на использовании гетерогенных катализаторов. Цеолитсодержащие катализаторы являются наиболее универсальными и перспективными из используемых контактов.
В процессе эксплуатации цеолиты, как и другие катализаторы процессов нефтепереработки и нефтехимии, постепенно теряют свою начальную активность. Время, в течение которого активность снижается до столь низкого уровня, что требуется замена катализатора или его регенерация, определяется типом процесса и условиями его проведения.
Правильно подобранные параметры процесса регенерации позволяют увеличить общий срок службы катализаторов, что позволяет снизить количество твердых токсичных отходов в виде отработанных контактов, скапливающихся на территории НПЗ и требующих утилизации. Помимо непосредственно вышедшего из строя катализатора, не малую антропогенную нагрузку на экосистемы прилежащих к предприятию территорий оказывает состав газов регенерации катализаторов, т.к. в их состав могут входить токсичные компоненты (CO, в малых количествах - окислы серы и азота).
Рентабельность новых процессов в значительной степени зависит от разработки наиболее дешевого и эффективного способа регенерации катализатора. Поэтому большое внимание исследователей уделяется выявлению причин дезактивации цеолитных катализаторов, поиску путей увеличения продолжительности работы катализаторов, технологии их регенерации [4]. Последняя является нестационарным процессом, и от условий ее проведения зависит, улучшатся или ухудшатся свойства катализатора после регенерации.
Процессы дезактивации катализаторов могут быть сведены к трем основным группам [5]: спекание (термическая дезактивация), отравление и блокировка. В процессах нефтепереработки катализатор дезактивируется в основном в результате блокировки активных центров коксовыми отложениями.
Дезактивация катализаторов блокировкой в отличие от дезактивации в результате отравления обычно является следствием отложения на их поверхности больших количеств коксогенов (чаще всего представляющих собой конденсированные ароматические или гибридные углеводороды, иногда с примесью металлоорганических соединений). Их масса может составлять 10-20 % от массы катализатора. В основном наблюдается два механизма блокировки.
Первый тип блокировки, в котором кокс образуется при определенных условиях в среде углеводородов. Поскольку эти отложения накапливаются вследствие протекания реакций крекинга и уплотнения исходных веществ, промежуточных и конечных продуктов, их полное устранение невозможно. Такой вид дезактивации можно минимизировать подбором технологических условий проведения процесса. При втором типе блокировки образуются отложения металлов в процессе переработки нефти (он встречается гораздо реже).
В некоторых случаях количество образуемого кокса может быть уменьшено путем введения в состав катализатора модифицирующих добавок.
Образование кокса, содержащего кроме углерода значительное количество водорода, а также следы серы, кислорода и азота, является наиболее распространенным механизмом блокировки поверхности катализаторов.
Однако блокировка поверхности может оказаться не единственной причиной дезактивации катализатора. Во многих процессах закоксовывание наблюдается одновременно с отравлением и со спеканием катализатора. Когда поверхность катализатора дезактивируется в результате отравления или блокировки, скорость реакции замедляется из-за того, что посторонние молекулы адсорбируются в порах катализатора и закрывают часть его активной поверхности. В результате сырье должно продиффундировать к недезактивированной части катализатора. Таким образом, дезактивация увеличивает среднее расстояние, на которое диффундирует реагент через пористую структуру [5].
Для цеолитов типа пентасил, термостабильность структуры которых очень велика, вероятнее всего, основной причиной дезактивации является отложение углеродсодержащих соединений на поверхности цеолита.
Схема образования кокса на поверхности цеолита представляет ряд последовательных реакций образования мономеров, уплотнения в результате нерегулярной конденсации и полимеризации с образованием циклов, т.е. связывание мономеров между собой и обеднением водородом вплоть до псевдографитовой структуры. Сам кокс в этом случае является смесью высокомолекулярных продуктов уплотнения, имеющий в своем составе последовательно переходящие друг в друга смолы, асфальтены и собственно кокс [6].
Характеристики коксовых отложений на катализаторе (химический состав, структура, дисперсность и распределение на поверхности) зависят от условий образования и могут изменяться в широких пределах. Заранее предсказать эти характеристики невозможно, их необходимо экспериментально определять в каждом конкретном случае. Полученные сведения открывают определенные возможности воздействия на процесс закокосовывания.
Предметом ряда исследований является выяснение вопроса о том, какие вещества или частицы можно считать предшественниками кокса – олефины или ароматические углеводороды. Большинство авторов полагают, что предшественниками являются ароматические соединения, хотя имеются доказательства в пользу важной роли в коксообразовании олефинов. Дать однозначный ответ на этот вопрос невозможно. Вероятнее всего, образование кокса на поверхности цеолитного катализатора может происходить из ароматических или олефиновых углеводородов через образование алкилароматических углеводородов с последующим превращением в полициклические соединения. При этом коксогенность углеводородов увеличивается с увеличением их молекулярного веса.
Большинство исследователей полагают, что высокая стабильность действия цеолитных катализаторов типа ZSM, обладающих необычными молекулярно-ситовыми свойствами, обусловлена структурными характеристиками, а медленная дезактивация – стерическими затруднениями образования кокса внутри кристаллического каркаса цеолита. Наряду с пространственно-селективными свойствами цеолитов ZSM-5 в процессе коксообразования большую роль играют кислотные центры его поверхности. Отложение кокса приводит к уменьшению силы и количества кислотных центров, что сопровождается снижением каталитической активности [6].
Дезактивация цеолитных катализаторов при закоксовывании может быть химической и химической и физической [7]. Химическая дезактивация является следствием химической адсорбции промежуточных продуктов уплотнения кокса. Такая адсорбция близка к случаям отравления ядами. Если реакции образования кокса протекают на тех же центрах, что и целевая реакция, они могут ее подавлять, т.к. продукты уплотнения иногда хемосорбируются и связываются с активными центрами сильнее, чем компоненты основного реагента.
По мере увеличения числа ядер в полиядерном ароматическом карбониевом ионе делокализация возрастает, что приводит к уменьшению целенаправленной реакционной способности переферийных атомов углерода конденсированной полиядерной системы. В результате постепенно исчезает сильное взаимодействие такой многоатомной системы с поверхностью катализатора, ослабевает химическое отравление продуктами уплотнения. На смену ему выступает физическое экранирование участков катализатора. Происходит закупорка каналов цеолита типа ZSM, сопровождающаяся выводом из строя основной части катализаторной поверхности.
Изучение коксовых отложений, которые образуются на разных катализаторах в условиях, отличающихся режимными параметрами и видом сырья, показывает, что они существенно отличаются по своим характеристикам [8]. Важнейшими из них при рассмотрении закономерностей окислительной регенерации являются химический состав кокса, его структура и дисперсность, а также распределение отложений по грануле катализатора.
1.3 Закоксовывание цеолитов в процессах переработки углеводородов
Исследование дезактивации цеолитов типа пентасил в процессах крекинга углеводородов проводится во многих странах. Полученные данные показывают [9], что в каналах H-ZSM-5 образуется мало углеродистых отложений. Кокс в основном формируется на внешней поверхности кристаллов цеолита и мало влияет на его активность. Закоксовывание H-ZSM-5 приводит к блокировке пор и отравлению кислотных центров. Скорость коксообразования тем выше, чем больше свободное пространство вблизи кислотного центра и чем ниже скорость десорбции продуктов-предшественников кокса. Сравнение спектров, полученных для образцов кокса, со спектрами графита и полиароматических соединений показало, что структура кокса сходна со структурами полиароматических соединений.
По данным [9], полученным при крекинге н-гексана, предшественниками кокса являются образующиеся в процессе олефины, которые подвергаются олигомеризации. В дальнейшем происходит циклизация олигомеров и образование за счет перераспределения водорода моноароматических молекул. Последние подвергаются алкилированию с последующей циклизацией алкильных фрагментов. Полиароматические молекулы (кокс) образуются в результате реакций перераспределения водорода.
В работе [9] приведены данные по изучению скорости дезактивации цеолита типа H-ZSM-5 и природа образующихся углеродистых отложений в зависимости от силикатного модуля цеолита, содержания модификатора и температуры реакции. В качестве модельной реакции использовали превращение гексена-1 в статической циклической системе в интервале температуры 398-523 К. Цеолиты с повышенным содержанием алюминия в решетке обладают не только повышенной активностью в начальный период процесса, но также замедленной дезактивацией, несмотря на более интенсивное углеобразование. Отложения, образующиеся при пониженных температурах в основном представляют собой насыщенные углеводороды. С увеличением соотношения Si/Al и времени контакта реагентов количество углерода в отложениях несколько увеличивается, причем с повышением температуры, и особенно в присутствии водяного пара эффект становится более явным.
Медленный линейный рост выхода кокса связан с низкой глубиной превращения н-гексана и небольшим количеством образующихся ароматических соединений, превращающихся в кокс. Здесь же методами ЭПР, рентгенографии, РЭС и ТПД-NH3
изучено влияние высокотемпературной обработки цеолита ZSM-5 парами TiCl4
на его каталитические свойства в реакции крекинга кумола и образование кокса. Обнаружено, что титан входит в решетку цеолита. С увеличением содержания титана уменьшается количество сильных кислотных центров и активность цеолита снижается. Но одновременно уменьшается коксообразование.
На свойства алюмосиликатных катализаторов сильно влияют накапливающиеся в них металлы. Отравление катализатора металлами может быть двух типов. Щелочные металлы нейтрализуют кислые центры катализатора и снижают в результате число работающих активных центров. Активность катализатора при этом снижается, но селективность остается неизменной. Отравление никелем, ванадием, железом, медью, свинцом мало влияет на кислотную активность катализатора, но приводит наряду с протеканием обычных реакций каталитического крекинга к катализу распада углеводородов на элементы, что резко увеличивает выход водорода и кокса. В наибольшей степени распад на элементы катализируют никель, кобальт и медь. Обычно сырье каталитического крекинга содержит железо, ванадий и никель. Отравляющее действие ванадия примерно в 4, а никеля — в 14 раз выше, чем железа. Помимо увеличения образования водорода и кокса, тяжелые металлы ускоряют спекание пор катализатора. Влияние отложения металлов на поверхности катализатора очень велико. Так, при повышении массового содержания никеля в катализаторе от 0,010 до 0,017% для сохранения выхода кокса оказалось необходимым понизить глубину крекинга в такой степени, что объемный выход бензина снизился с 48,9 до 43,9 % на сырье; при этом выход водорода повысился в 6 раз [9].
Важной особенностью пентасилов, отличающей их от других цеолитов, является высокая стабильность каталитической активности при проведении высокотемпературной ароматизации, т.е. очень слабое влияние образующегося кокса на каталитическую активность цеолита. Возникающие в процессе ароматизации или введенные в цеолит путем предварительной адсорбции ароматические молекулы не образуют в каналах цеолитов этого типа (в отличие от НМ) продуктов уплотнения как в ходе реакции, так и при последующей термообработке. Однако на внешней поверхности пентасила существуют центры, катализирующие образование слабо графитированных продуктов уплотнения [10,11]. Отсутствие кокса внутри каналов пентасила подтверждается следующими данными [10]:
-стабильной активностью НЦВК при высокотемпературной ароматизации олефинов;
-сохранением исходной ароматизирующей активности у образца, отравленного 2,4,6-триметилпиридином и содержащего адсорбированный п-ксилол, после термической обработки на воздухе при температуре 300 0
С;
- неизменностью ароматизирующей способности образца, выгруженного после проведения реакции ароматизации изобутилена при температуре 320 0
С и подвергнутого термоокислительной обработке на воздухе при 300 0
С.
Существенное отличие НЦВК от НМ в том, что образующиеся на внешней поверхности пентасила слабографитированные продукты уплотнения уже при низкотемпературной (300 0
С) окислительной обработке превращаются в сильно графитированные структуры. При этом такие структуры, локализованные на внешней поверхности катализатора, не влияют не только на идущую внутри каналов ароматизацию, но и на протекающие на внешней поверхности низкотемпературные превращения олефина. Считают, что эта реакция катализируется поверхностными кислотными центрами цеолита.
Таким образом, ароматические углеводороды, образующиеся на пентасилах при высокотемпературных превращениях олефинов, слабо связаны в каналах цеолита и не образуют в них продуктов уплотнения. Причиной этого явления является малый диаметр каналов, геометрически ограничивающий возможность образования поликонденсированных молекул и малая плотность активных центров.
Показано [9], что скорость дезактивации катализатора в результате зауглероживания зависит от плотности кислотных центров, размера пор и температуры реакции. Имеются данные, что дезактивация цеолита типа ZSM в процессах алкилирования снижается в случае прессования образцов при повышенном давлении (108
Па). Этот эффект, по-видимому, связан с уменьшением свободного межкристаллитного пространства, что затрудняет его прямой контакт с внешней газовой фазой и ограничивает роль частиц-предшественников кокса.
Там же представлены данные, полученные при изомеризации гексена-1 на H-ZSM5 при температуре 200-280 0
С. Основными продуктами являются цис- и транс-гексен-2. Все продукты скелетной перегруппировки являются вторичными. Доля продуктов крекинга и полимеризации при температуре 200 0
С на ZSM-5 заметно меньше, чем на HY. Найдено, что кокс является первичным продуктом, его образование снижается с ростом температуры. Анализ соотношения С/Н показывает, что на ZSM-5 кокс состоит в основном из олефинов.
1.4 Механизм образования и состав кокса
Химический состав кокса, отлагающегося на катализаторе, определяется в первую очередь механизмом его образования [12]. В настоящее время выделяют два механизма: консекутивный и карбидного цикла. Согласно консекутивной схеме, отложения кокса на поверхности катализатора формируются в результате протекания последовательных реакций нерегулярной конденсации и полимеризации углеводородов, сопровождающихся возникновением и связыванием циклических структур. При этом наблюдается их постепенное обеднение водородом вплоть до псевдографитовой структуры за счет выделения легких углеводородов и водорода. Сам кокс в этом случае представляет собой смесь высокомолекулярных продуктов уплотнения от смол и асфальтенов до карбоидов и в предельном случае - до графитоподобных отложений. Истинный химический состав такой смеси определить практически невозможно, поэтому состав кокса принято характеризовать усредненным элементным составом.
Результаты изучения состава кокса, отлагающегося на катализаторах различных процессов, указывают на наличие в нем, кроме углерода, и водорода. По мнению некоторых исследователей, углерод в обычных условиях каталитических процессов вообще не способен существовать в чистом виде, а он всегда входит в состав полициклических ароматических углеводородов, в которых доля водорода определяется степенью конденсации и может быть сколь угодно малой. При образовании кокса по консекутивному механизму отношение Н/С зависит от типа катализатора, состава перерабатываемого сырья, температуры, времени проведения процесса, степени закоксованности и условий последующей продувки закоксованного катализатора. Для одного и того же катализатора отношение Н/С в отложениях кокса может изменяться в широких пределах при изменении состава перерабатываемого сырья. С повышением температуры кокс уплотняется за счет отщепления и удаления легких углеводородов. Наблюдается непрерывный рост молекулярной массы, доли ароматических структур, уменьшение отношения Н/С.
Кокс, образующийся на катализаторах по консекутивному механизму, не однороден по своему составу. Наряду с углеродом в нем содержится водород, а в некоторых случаях присутствуют и сера, и кислород. Неоднородность состава и структуры кокса обусловливают его неодинаковую реакционную способность к окислению. При термическом анализе образца алюмохромового катализатора, закоксованного при обработке парами н-пропилового спирта, на кривой изменения температуры наблюдаются два максимума [13]. Первый максимум отмечен при 285 °С, а второй, отвечающий, по мнению автора, второй составляющей кокса, приходится на 370-380 °С. В дальнейшем две отчетливо различные области выгорания кокса были обнаружены и на других закоксованных катализаторах [14, 15]. Наличие двух областей может быть обусловлено несколькими причинами, например, неоднородностью кокса по химическому составу. Так, в одной из первых работ по изучению кинетики окислительной регенерации катализаторов крекинга наблюдали связь между скоростью выгорания кокса и его составом [16]. Исследования проводили в интервале температур 500-610°С на образцах катализатора с различным содержанием, кокса. Во всех экспериментах отмечено преимущественное выгорание водородсодержащих компонентов в начальные моменты. В дальнейшем эти результаты были неоднократно подтверждены.
1.5 Процессы окисления кокса.
Рассмотрим современные представления о процессе окисления углерода в интервале температур, характерном для окислительной обработки закоксованных катализаторов - от 150 до 700 °С.
В общем случае горение углерода характеризуется следующими процессами [14, 15]:
-химическое взаимодействие углерода с кислородом или собственно горение углерода, в результате которого образуются оксиды углерода:
С + О2
= СО2
+ 395,4 кДж/моль;
С + 0,5О2
= СО + 110,4 кДж/моль;
-дальнейшие превращения образующихся оксидов:
СО + 0,5О2
= СО2
+ 285,0 кДж/моль;
С + СО2
= 2СО - 172,2 кДж/моль;
-взаимодействие углерода с водяным паром, поскольку при окислении угля практически всегда в реакционной зоне присутствуют пары:
С + Н2
О = СО + Н2
+ 41,0 кДж/моль.
Однако до 700 °С реакциями непосредственного взаимодействия углерода с парами воды и диоксидом углерода можно пренебречь и характеризовать окисление только процессами взаимодействия с кислородом и доокисления оксида углерода в газовой фазе [17]. В то же время необходимо отметить, при температурах ниже 750 °С скорости окисления углерода в сухой среде весьма малы.
По аналогии с горением угля можно условно выделить две составляющие кокса - легкогорючую и трудногорючую. Первая состоит в основном из аморфной, наиболее богатой водородом части кокса, а трудногорючая составляющая включает высококонденсированные псевдографитные структуры. Соотношение этих составляющих, как уже отмечалось, в первую очередь определяется температурой каталитического процесса и условиями последующего отпаривания и продувки инертным газом [16].
Регенерация зерна катализатора путем выжигания выделившегося углеродсодержащего кокса в порах соответствует специальному случаю некаталитических реакций газ - твердая фаза [5]. Существует обширная литература по некаталитическим процессам, отражающая их промышленную важность, поскольку такие процессы включают восстановление железной руды, обжиг известняка, сжигание твердого топлива и т. д. Процесс регенерации это специальный случай сгорания, в котором топливо (кокс) размещено внутри пористой матрицы. В результате реакции углерод удаляется из матрицы путем окисления и перехода в газообразные продукты. Таким образом, пористая структура катализатора существует как резервуар, в котором кокс выделяется и из которого он удаляется; в идеальном процессе пористая структура должна оставаться неизменной. В этом отношении данный процесс отличается от таких реакций, как сжигание кусков угля, где пористая структура постоянно изменяется во время протекания процесса. Второе отличие от ряда других некаталитических процессов заключается в том, что во многих из них достаточен изотермический анализ, так как, хотя реакция может быть неизотермической, конверсия может быть предсказана достаточно точно для целей расчета на основе любой изотермической модели. Когда катализаторы регенерируются путем выжигания кокса, кроме скорости конверсии (время, требующееся для регенерации) важно задать максимальную температуру во время процесса, чтобы защитить катализатор от спекания. Таким образом, при регенерации катализатора важным является проявление неизотермичности, даже если все остальные требования к процессу выполнены.
В общем случае механизм регенерации отдельного зерна катализатора очень сильно зависит от используемой температурной области [5], как показано на рисунке 1.1.
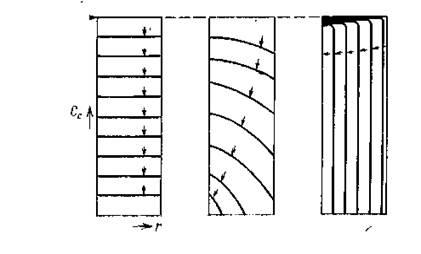
Рисунок 1.1 -
Профили концентрации кокса в сферических частицах катализатора для последовательных стадий выжигания: а
— низкая температура; б
— промежуточная температура; в
— высокая температура
При низких температурах кислород имеет доступ ко всем местам выделения кокса по всему объему зерна; при этом протекает реакция газ - твердая фаза гомогенного типа, подобно реакциям в обычных каталитических процессах, протекающих при значениях фактора эффективности, близких к единице. В другом предельном случае, при высоких температурах, скорость окисления становится очень большой, так что реакция начинает лимитироваться массопереносом кислорода через поры. При этих условиях действует механизм нарастающей оболочки, как показано на рисунке 1.1(в).
Ранние работы по регенерации зерен относились в основном к изотермической задаче и заключались в оценке времени, требующегося для полного удаления всего кокса из зерна. В одной из них были определены два четко различающихся периода по скорости процесса: период постоянной и период падающей скорости. Первый соответствовал выжиганию кокса на поверхности. Период падающей скорости относится к выжиганию кокса внутри зерна; профили концентрации кислорода были получены также и для этого случая, но выражение для скорости в этом периоде является сложной функцией доли оставшегося кислорода.
1.6 Изменения структуры катализатора в процессе его регенерации.
Со временем катализаторы стареют, что проявляется в снижении каталитической активности. Старение катализаторов обычно вызывается рекристаллизацией и связанными с ними изменениями структуры поверхности.
Во многих случаях дезактивация наступает по другим причинам. Например, поверхность катализатора может покрываться пленкой смолообразных веществ и отложений угля в результате побочных реакций, в данном случае утомленным катализаторам регенерацией можно вернуть полностью или частично их исходную активность.
Методы регенерации очень разнообразны и специфичны для отдельных катализаторов. Такие термически стабильные катализаторы, как Al2
О3
,. Th02
, ZnO, Cr2
O3
, алюмосиликаты и т. д., регенерируются прокаливанием в токе воздуха или кислорода для выжигания из них посторонних веществ, содержащих углерод. Регенерацию катализаторов следует вести при строго контролируемой температуре, так как в противном случае они могут потерять часть своей активности из-за укрупнения кристаллов в результате перегрева. Во избежании опасного повышения температуры часто необходимо разбавлять газы азотом или парами воды. Никелевые и кобальтовые катализаторы регенерируют окислительно-восстановительным методом, который заключается в осторожном окислении катализатора воздухом с последующим восстановлением образующихся окислов.
Алюмосиликатые катализаторы для каталитического крекинга, теряющие через 10 минут свою активность из-за отложения в них кокса, полностью регенерируют горячим воздухом. Активные медные катализаторы регенерируют повторным пропусканием водорода при 180-200°С.
Существуют и другие методы регенерации: экстракция растворителями, протравление поверхности катализаторов кислотами или щелочами, переосаждение и т. п. [39].
Цеолитсодержащие катализаторы в основном для восстановления их активности подвергают окислительной регенерации. Целевое назначение процесса окислительной регенерации - удаление кокса без ухудшения свойств катализатора. На практике достичь этого не удается, так как окислительная среда, присутствие в газе паров воды и интенсивное выделение тепла при горении кокса оказывают определенное воздействие на катализатор. В ряде случаев изменения незначительны, однако нередко активность и селективность свежего и регенерированного катализаторов различаются существенно [16]. Это происходит из-за изменения химического состава катализаторов, сопровождающегося изменением удельной активности, и вследствие структурных и других превращений, приводящих к изменению удельной поверхности или ее доступности.
При обсуждении вероятного механизма окисления кокса на катализаторах отмечалось, что последние могут служить переносчиком кислорода из газовой фазы к коксу по стадийному механизму. И если лимитирующей стадией является присоединение кислорода к катализатору, он существует в начальные моменты окислительной регенерации в восстановленной форме. Окисление компонентов катализатора в этом случае может протекать в основном после выжига кокса и затрагивать только поверхность катализатора. Если же лимитирующей стадией является передача кислорода коксу от катализатора, последний будет быстро окисляться. При этом окислению, по-видимому, будут подвергаться не только поверхностные слои, но и объем катализатора. Получающийся при регенерации оксид активного компонента катализатора в определенных условиях может взаимодействовать с носителем с образованием соединений, не обладающих каталитической активностью.
Промышленные катализаторы, как правило, представляют собой системы, по многим параметрам далекие от термодинамического равновесия. Это обусловлено развитой поверхностью и наличием микроискажений решетки кристаллов. При низких температурах неравновесное состояние высокодисперсной структуры может сохраняться весьма длительное время. С повышением температуры увеличивается подвижность элементов структуры твердого тела, и система стремится перейти в более устойчивое состояние. Поэтому практически все промышленные катализаторы в процессе эксплуатации (особенно на стадии регенерации) постепенно претерпевают структурные изменения. В большинстве случаев уменьшается удельная поверхность, происходит перераспределение объема пор по радиусам, и чаще всего размер пор возрастает, а общая пористость катализаторов уменьшается. Необходимо отметить, что для сложных катализаторов кроме изменения структуры в объеме гранул возможно изменение соотношения площадей поверхности (дисперсности) различных фаз [21].
Изменения пористой структуры и поверхности обусловливаются двумя процессами: кристаллизацией и спеканием. При кристаллизации катализаторов имеет место рост кристаллов и упорядочение всей структуры с устранением дефектов и других искажений в решетке кристаллов. В результате исчезают наиболее мелкие частицы, увеличивается размер пор, сокращается удельная поверхность. Однако общий объем пор при этом изменяется незначительно. В процессе кристаллизации формируется относительно стабильная и более однородная структура.
Спекание - это процесс беспорядочного уплотнения системы, сопровождающийся уменьшением удельной поверхности и объема пор. Формирующаяся при спекании структура мало устойчива, она склонна к дальнейшему уплотнению и кристаллизации. Механизмы кристаллизации и спекания различны [21]. Кристаллизация обусловлена преимущественно поверхностной диффузией, а при спекании большую роль играет объемная диффузия, при которой первичные частицы просто срастаются друг с другом. Кристаллизация и упорядочение структуры протекают в области более низких температур, чем спекание.
В отличие от кристаллических пористых тел избыточная свободная энергия аморфных структур определяется лишь величиной удельной поверхности. Поэтому спеканию аморфных тел не предшествуют процессы упорядочения структуры [22].
Для большинства катализаторов переменной активности наибольшее изменение пористой структуры и поверхности наблюдается при окислительной регенерации, так как она проводится при более высоких температурах, чем основной каталитический процесс [21]. Ускорение спекания вызывается перегревом закоксованных частиц или наиболее закоксованных их участков в процессе регенерации. Перегревы могут достигать сотен градусов. При этом наряду со спеканием в некоторых случаях происходит растрескивание катализатора.
Другим фактором, ускоряющим процесс спекания при регенерации, может быть воздействие паров воды при высокой температуре.
Неодинаковое изменение структуры, например, алюмосиликатных катализаторов после прокаливания при высокой температуре и обработки водяным паром отмечалось разными исследователями. Исследование структурных изменений и выявление закономерностей, по которым они протекают в результате прокаливания и обработки паром, было проведено на образце алюмосиликатного катализатора [16]. Показано, что и перегрев до 900 °С на воздухе, и обработка паром при 750 °С приводят к уменьшению поверхности катализаторов и объема пор. Отмечено различное действие термической и паровой обработок на пористую структуру. Так, при перегреве катализатора в воздухе удельная поверхность уменьшается приблизительно пропорционально сокращению объема пор. Размеры пор существенно не меняются. В случае же обработки паром объем пор сокращается значительно медленнее, чем удельная поверхность, при этом размеры пор резко возрастают. При длительном воздействии пара при температуре 750 °С полностью исчезают мелкие поры, катализаторы становятся крупнопористыми. Кроме того, водяной пар ускоряет уменьшение удельной поверхности катализатора.
Представляют интерес и результаты исследований отдельных частиц шарикового катализатора [16, с.56]. Образцы были отобраны из регенераторов промышленных установок. Частицы равновесного катализатора - «черные», «серые» и «белые» шарики одинакового размера (
d
=
= 3-4 мм) существенно отличаются друг от друга. «Белые» шарики - те, с которых кокс полностью выгорел в регенераторе промышленной установки. «Черные» и «серые» содержат кокс во всем объеме частицы, хотя они и прошли через регенератор. Из них были приготовлены пластинки толщиной 0,1-0,2 мм. «Черные» и «серые» пластинки регенерировали в муфельной печи при 700°С. Через каждые 5-10 минут регенерации с помощью микроскопа измеряли границы той части пластинки, которая посветлела в результате регенерации.
«Серые» пластинки весьма неоднородны. Одни из них регенерируются быстро и посветление происходит почти одновременно по всей закоксованной зоне. На других обнаруживаются зоны с разной скоростью удаления кокса.
Когда разница в скорости регенерации зон достигает еще большей величины, в некоторый момент времени наблюдается закоксованное кольцо. И, наконец, у части пластинок после первоначального уменьшения диаметра закоксованного ядра границы его стабилизируются и не изменяются длительное время (10 часов). По-видимому, этот кокс вообще не удаляется с катализатора.
Пластинки, полученные из «черных» шариков, регенерируются поэтапно. Вначале от кокса освобождается одна из зон пластинки, она светлеет одновременно. Затем длительное время границы закоксованной части практически не меняются, пока не посветлеет новая зона. Чаще всего первой регенерируется зона, расположенная в центре пластинки, а затем зоны, примыкающие к предыдущей.
Внешнее периферийное кольцо обычно не светлеет даже при очень длительной регенерации. Подобным образом выжигается большинство пластинок.
С образованием закоксованных колец некоторые пластинки не светлеют даже частично, несмотря на длительную регенерацию при температуре до 750 °С. Таким образом, в объеме одного шарика может быть несколько участков, которые освобождаются от кокса в разное время.
Различная длительность выгорания кокса с отдельных зон катализатора обусловлена неравной их закоксованностью или неодинаковыми физико-химическими свойствами. В дальнейшем было установлено, что концентрация кокса по глубине частицы действительно может меняться значительно и при некоторых условиях не плавно, а скачкообразно. Разная концентрация кокса неизбежно приводит и к различной длительности полного его выгорания при регенерации пластин катализатора. Но этим не объясняется абсолютная невозможность регенерации некоторых зон катализатора при жестких условиях на тонких пластинках. Такой кокс можно представить лишь как заплавленный в вещество катализатора при полном спекании данного участка частицы.
Последнее подтверждается невозможностью регенерации основной массы целых, не имеющих повреждений поверхности «черных» шариков даже при 750 °С в среде кислорода в течение 20 часов. В то же время центральная часть пластинок, изготовленных из таких шариков, обычно освобождалась от кокса уже при 700 °С в воздухе за 10-60 мин. Следовательно, «черные», нерегенерируемые частицы окружены плотной спекшейся коркой, закрывающей доступ кислорода внутрь.
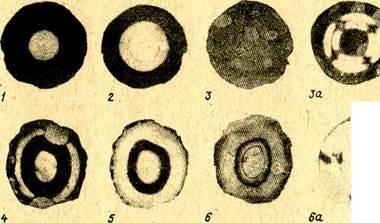
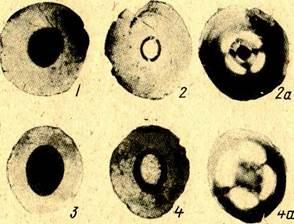
Была предпринята попытка охарактеризовать пористую структуру отдельных зон катализатора [23]. Внутренние зоны катализатора, освобождающиеся от кокса в разное время, отличаются оптическими свойствами. На рисунках. 1.2 и 1.3 приведены фотографии пластинок из «серых» и «черных» частиц в проходящем свете в разные моменты регенерации. На фотографиях, выполненных в обычном свете, можно различить границы некоторых зон катализатора. Отчетливы они у пластинок из «черных» частиц. Особенно четко границы видны на фотографиях, сделанных в поляризованном свете. Если на пути поляризованного луча нет кристаллического вещества, на фотографиях изменений нет. Если же на пути луча имеется кристалл, то вследствие двойного лучепреломления соответствующие участки катализатора на фотографиях выглядят светлыми. По интенсивности посветления можно приближенно судить о концентрации кристаллического вещества. Из рисунка 1.3 видно, что в «белых» частицах кристаллическая фаза отсутствует. Более сильное посветление наблюдается в отдельных зонах регенерированных «серых» пластинок. Регенерированная часть «черных» пластинок также содержит несколько зон с кристаллической фазой, концентрация которой обычно больше, чем у зон «серых» пластинок. От центра к периферии интенсивность посветления зон обычно увеличивается вплоть до границ остаточного кокса у «серых» частиц или до нерегенерируемой спекшейся корки у «черных» частиц. Имеются и такие частицы, у которых светлые и темные области чередуются.    Ядра играют незначительную роль в процессе каталитического крекинга. В то же время результаты анализа ядер после регенерации свидетельствуют о том, что, обладая довольно большой удельной поверхностью, в два с лишним раза превышающей удельную поверхность периферийных слоев катализатора, они могли бы эффективно участвовать в процессе. Ядра играют незначительную роль в процессе каталитического крекинга. В то же время результаты анализа ядер после регенерации свидетельствуют о том, что, обладая довольно большой удельной поверхностью, в два с лишним раза превышающей удельную поверхность периферийных слоев катализатора, они могли бы эффективно участвовать в процессе.
Таким образом, в промышленных условиях катализатор может спекаться по глубине шарика неравномерно, поэтому структура по диаметру частицы неоднородна. Эта неоднородность увеличивается при переходе от «белых» к «серым» и от «серых» к «черным» шарикам.
Рисунок 1.2 - Пластинки, приготовленные из «черных» шариков: 1-6-в обычном свете; 3а, 6а-в поляризованном свете
Известны случаи быстрого (в течение нескольких часов) спекания аморфного алюмосиликатного катализатора на установках крекинга с псевдоожиженным слоем. Современные марки цеолитсодержащих катализаторов эксплуатируют длительное время при температурах до 760°С без существенного снижения их каталитических свойств.
 
Рисунок 1.3 - Пластинки, приготовленные из «серых» и «белых» шариков: 1,3 - исходные «серые» пластинки; 2, 4 - то же, в конце регенерации; 2а, 4 - то же, в поляризованном свете; 5-7
- «белые» пластинки в поляризованном свете
Катализаторы других процессов менее термостабильны. Установлено, что действие температур на уменьшение поверхности алюмохромовых катализаторов начинает сильно сказываться выше 640-650 °С [16]. Для нанесенных катализаторов при окислительной регенерации может наблюдаться уменьшение дисперсности активного компонента. Основной причиной изменения дисперсности активного компонента в нанесенных катализаторах, как и в случае других пористых катализаторов, является удаленность системы от состояния равновесия [21]. После периода разработки дисперсная структура катализатора находится в некотором стационарном состоянии, когда дисперсность в данных температурных условиях не изменяется. Однако в процессе окислительной регенерации перегревы и действие паров воды ускоряют рост частиц. Например, под действием высоких температур происходит укрупнение частиц платины на поверхности носителя.
При нагревании до 500 °С наблюдается рост частиц платины и соответствующее уменьшение поверхности платины и степени превращения в реакции гидрирования бензола. При нагревании до 600-800 °С платиновый катализатор практически полностью теряет активность.
Природа носителя также оказывает непосредственное влияние на стабильность структуры катализаторов в процессе их регенерации. Так, в [21] показано, что стабильность структуры катализаторов при нагревании зависит не только от физических свойств активного материала, но и от природы носителя. Вследствие разной поверхностной подвижности атомов на различных поверхностях природа носителя влияет и на размеры, и на форму дискретных частиц активного компонента, образующихся при нагревании контактов. Кристаллизация одного и того же вещества на разных носителях приводит к формированию структур, различающихся как внешней формой кристаллов, так и их размерами.
Влияние природы носителя на поверхностную миграцию активного компонента было отмечено и в других работах. При исследовании термостабильности никеля в различных катализаторах (никельхромовом, никельалюминиевом, никельхромалюминиевом) установлено [16], что скорость миграции никеля по поверхности зависит и от температуры, и от прочности связи частиц никеля с поверхностью. Эта величина, как полагают авторы, зависит от прочности связи никеля с носителем. Наблюдаемое с ростом прочности связи увеличение термостойкости поверхности никеля указывает на снижение скорости диффузии по поверхности.
Таким образом, изменения структурных характеристик или размеров нанесенного на носитель активного компонента проявляются у всех катализаторов. Спекание может протекать по разным механизмам и в зависимости от условий регенерации и свойств катализатора может вызывать кристаллизацию вещества катализатора. В связи с этим при изучении спекания катализатора в конкретном процессе необходимо прежде всего выяснить, какой из возможных механизмов играет большую роль, что позволит наметить пути повышения стабильности катализатора.
1.7 Способы увеличения стабильности цеолитных катализаторов
Актуальной в современных процессах нефтепереработки является проблема придания стабильности цеолитсодержащему катализатору, что позволит увеличить сроки его межрегенерационного пробега и тем самым уменьшить капиталозатраты в производство. С этой целью прибегают к различным методам модификации цеолитов. Так авторы [24,25] утверждают, что стабильность каталитического действия цеолитов зависит от числа атомов алюминия в катионных позициях. Методами ЯМР, 27
AL, ИКС, РФЭС ими исследованы серии цеолитов типа Y и ZSM, подвергнутые деалюминированию парами SiCl4
. Показано, что термохимическая обработка цеолитов Y и ZSM парами SiCl4
приводит к удалению в первую очередь атомов Al кремнекислородного каркаса цеолита. Активные центры, в состав которых входят атомы алюминия катионных позиций, в значительной степени ведут реакции перераспределения водорода в промежуточных ненасыщенных углеводородах. С ростом содержания атомов алюминия в катионных позициях стабильность уменьшается. Неравномерность распределения атомов алюминия по кристаллоцеолитам повышает вероятность образования фрагментов типа: [(SiO4
)4-
n
Si(OAl)n
]n
-
Al3+
или [(SiO4
)4-
n
Si(OAl)n
]n
-
Al3+
(OH-
…H+
), которые могут являться центрами многоточечного взаимодействия с молекулами реагентов.
В работе [26] выявлена зависимость направления протекания превращений н-бутана от температуры на цинкмодифицированном цеолите типа ЦВМ. Установлено, что пропиленсодержащую фракцию процесса дегидрирования бутана можно переработать по двум направлениям:
- получение высокооктановых компонентов моторных топлив с низким содержанием ароматических углеводородов. Процесс рекомендуется вести при 300-330 0
С, объемной скорости 300-720ч-1
, продолжительность до 30ч.
- получение концентрата ароматических углеводородов, который может быть использован в качестве высокооктановой добавки к моторным топливам, либо как сырье для нефтехимии. Процесс рекомендуется проводить при 500-530 0
С, объемной скорости 300-720ч-1
, продолжительности до 20 часов.
Имеются также варианты повышения активности цеолитных катализаторов на стадии их синтеза [27]. Способ получения микросферического цеолитсодержащего катализатора для крекинга нефтяных фракций включает осаждение из водных растворов алюмината натрия и сернокислого алюминия аморфной гидроокиси алюминия, смещения водной суспензии гидроокиси алюминия в количестве 5-40% с алюмосиликатной суспензией и цеолитом с последующей распылительной сушкой. Повысить срок службы катализатора можно введением в состав цеолита железа [28]. Срок службы декатионированного цеолита в этом случае увеличивается с 85 до 570 ч для образца с содержанием железа 2,26%. Дальнейшее повышение концентрации железа в цеолите снижает срок службы катализатора.
Зависимость кислотных свойств цеолита от концентрации вводимого железа обуславливает и изменение его каталитических свойств. Введение железа на стадии гидротермального синтеза значительно повышает селективность катализатора по отношению к олефинам С2
-С4
. Это объясняется снижением активности Fe-содержащих катализаторов во вторичных реакциях, к числу которых относится, например, реакция перераспределения водорода. Обнаружено промотирующее влияние железа (III) при нанесении его на декатионированный и деалюминированный высококремнистый цеолит типа пентасил в реакциях изомеризации дихлорбензолов и их алкилирования этиленом [28].
В процессе циклоформинга бензинового сырья в [29] использовали цинк-модифицированный цеолитсодержащий катализатор, в котором варьировали содержание цинка от 0 до 10 %. Полученные результаты согласуются со схемой образования АРУ из низкомолекулярных олефиновых продуктов крекинга, предложенной в [30] для СВК-цеолитов. Модифицирование катализаторов цинком способствует превращению продуктов крекинга в ароматические компоненты, как и при введении галлия в цеолиты типа ZSM [31], и протеканию реакций дегидрирования углеводородов, на что указывает непрерывное возрастание выхода водорода.
Наличие в составе катализатора металла - катализатора окислительно-восстановительных реакций - позволяет значительно снизить содержание кокса в регенерированном катализаторе - до 0,1% и менее, так как скорость горения остаточного кокса возрастает в этом случае на порядок и более [32].
В качестве металлов-промоторов, интенсифицирующих регенерацию закоксованного катализатора, применяют чаще всего платину, нанесенную в малых концентрациях (< 0,1 % масс.) либо непосредственно на цеолитсодержащий катализатор, или на окись алюминия с использованием как самостоятельной добавки к каталитической композиции [34]. Это позволяет значительно повысить полноту и скорость сгорания кокса катализатора и существенно понизить содержание монооксида углерода в газах регенерации, тем самым предотвратить неконтролируемое загорание СО над слоем катализатора в регенераторе, приводящее к прогару циклонов, котлов-утилизаторов и другого оборудования. Из отечественных промоторов окисления можно отметить КО-4, КО -9, Оксипром-1 и Оксипром-2.
Помимо непосредственного модифицирования состава катализатора используют и другие методы, позволяющие повысить срок активной работы контакта. Для снижения дезактивирующего влияния примесей сырья на цеолитсодержащий катализатор также весьма эффективно применяют технологию каталитического крекинга с подачей в сырье специальных пассиваторов металлов [34]. Последние представляют собой металлоорганические комплексы сурьмы, висмута, фосфора или олова. Сущность эффекта пассивации заключается в переводе металлов, осадившихся на катализаторе, в пассивное состояние, например, в результате образования соединения типа шпинели. Пассивирующий агент вводят в сырье в виде водо- или маслорастворимой добавки. Подача пассиваторов резко снижает выход кокса и водорода, увеличивает выход бензина и производительность установки. В настоящее время пассиваторы применяют на 80 % установок каталитического крекинга остатков в США и около 50 % установок в Западной Европе.
В последние годы внедряется ЦСК с твердой добавкой - ловушкой ванадия и никеля, содержащей оксиды Са, Mg, титанат бария и др., адсорбирующие в 6-10 раз больше металлов, чем сам катализатор.
Ну и, наконец, технологические параметры проведения процесса также оказывают непосредственное влияние на коксообразование и, следовательно, на время стабильной работы катализатора. Поэтому смягчение технологических режимов процессов приведет к повышению срока службы катализатора.
1.8 Способы осуществления регенерации в промышленности
Одной из определяющих эксплуатационных характеристик промышленных катализаторов является их регенерируемость. Цеолитсодержащие катализаторы имеют несколько лучшие регенерационные характеристики, чем аморфные алюмосиликаты. Применение в цеолитсодержащих катализаторах редкоземельного цеолита улучшает регенерацию вследствие катализирующего действия ионов редкоземельных элементов на горение кокса [35].
Регенерация катализатора обычно значительно сложнее, чем проведение самого процесса [36]. Сущность регенерации заключается в сгорании коксовых отложений при их контактировании с кислородом воздуха. В результате выделяется значительное количество тепла (от 6000 до 7500 ккал/кг кокса), которое необходимо частично отводить из зоны регенерации, чтобы не перегреть всю массу катализатора. При этом продолжительность регенерации не должна быть чрезмерно большой, чтобы регенератор был приемлемых размеров. Регенерационная способность катализатора - скорость выжига кокса, выраженная в г/(л-ч), но обычно - в кг кокса с 1 т катализатора в час, равная 50-80 кг/(т-ч).
Современный процесс, протекающий с дезактивацией катализатора, может быть эффективным лишь в том случае, если обеспечивает возможность простого осуществления регенерации катализатора. Поэтому в разных странах ведутся исследования с целью определения оптимальных условий реакции. В промышленных условиях для удаления кокса наиболее широко используется окислительная регенерация – процесс контролируемого выжига кокса кислородсодержащими смесями при температуре катализа и выше [37].
Окислительная регенерация представляет собой совокупность химических реакций, протекающих при взаимодействии кислорода с коксом, в результате которых кокс удаляется в виде газообразных продуктов. Выжиг кокса можно интенсифицировать, повышая содержание кислорода в газе и температуру регенерации, а также путем введения в состав катализатора промоторов окисления, которые не оказывают заметного влияния на его активность и селективность. При повышении температуры регенерации необходимо учитывать возможность спекания и растрескивания катализатора, так как целевым назначением процесса окислительной регенерации является удаление кокса без ухудшения свойств катализатора.
Регенерация катализаторов ведется горячим воздухом при температуре 650-750 °С, причем эта температура регулируется количеством дутья при коэффициенте избытка воздуха 1. При этом часть кокса сгорает до СО2
(теплота сгорания 33 МДж/кг), а остальной кокс - до СО (теплота сгорания 10 МДж/кг). Обычно в продуктах горения кокса мольное соотношение СО:СО2
равно примерно 1:1. В закоксованном катализаторе содержится 1,2-2,0 % (мас.) кокса, а после регенерации - не более 0,1 % (стремятся к 0,05 %) [38].
Регенерацию катализатора, как правило, не доводят до конца: частица отрегенерированного катализатора состоит обычно как бы из темного ядра, где в самых глубоких порах катализатора остается так называемый «глубинный» или остаточный кокс, и светлой отрегенерированной оболочки [36]. Остаточный кокс может составлять от 0,1 до 0,8% в пересчете на катализатор. Желательны значения остаточного кокса не более 0,1 %,
так как повышенные его количества снижают начальную активность катализатора, а также способствуют его разрушению при колебаниях температуры.
Конструкция регенератора в значительной степени определяется тем, в каком реакторном аппарате проводится основной процесс. Для процессов со сплошным движущимся или псевдоожиженным слоем катализатора реализуется сменно-циклический режим работы [37]. При этом регенерацию проводят непрерывно в отдельном аппарате, так же как процесс в реакторе (т.е. в движущемся или псевдоожиженном слое). Напротив, для аппарата с неподвижным слоем катализатора реализуется, как правило, сменно-циклический режим работы: основной процесс и регенерация проводятся последовательно в одном и том же аппарате.
При регенерации неподвижного слоя катализатора важно выдерживать такие условия, чтобы максимальная температура в зоне горения не превышала значения, при котором дезактивируется катализатор. Многие опубликованные работы по регенерации неподвижных слоев катализатора рассматривают явление повышения температуры. Это связано с влиянием повышения температуры на каталитическую активность и селективность. Повышение температуры является функцией величины температуры и координаты точки в слое; и можно определить три различных максимума температуры. Это - максимум температуры в данной точке слоя в любой момент времени в течение регенерации; максимум температуры в данный момент времени в пределах любой части слоя; наконец, пик температуры, который можно определить как наибольшую температуру, достигаемую в течение всего процесса в любой точке слоя в любой момент времени [2].
Повышение температуры, развивающееся в адиабатическом неподвижном слое в период регенерации, достигает максимума в начальных стадиях процесса, когда отрегенерирована только небольшая часть слоя. Это связано с высокой начальной скоростью реакции, возникающей при контактировании относительно высококонцентрированного газа с закоксованным катализатором. Температурный пик быстро достигает асимптотически максимального значения для оставшегося периода процесса, лимитируемого диффузией. Это асимптоматическое максимальное значение зависит как от начальных концентраций кислорода, так и от начальных концентраций.
Одна из возможностей, которую следует рассмотреть, заключается в резких скачках температуры. Зона сгорания (или реакционная зона) движется вдоль слоя со скоростью, определяемой скоростью реакции и начальными значениями концентраций азота и кислорода. Результирующий тепловой фронт, вызванный экзотермичностью процесса сгорания кокса, движется независимо от зоны сгорания из-за конвективного влияния газового потока. Если зона сгорания и тепловой фронт перемещаются с одинаковой скоростью, то тепло реакции накапливается в пределах зоны сгорания и температура этой зоны начинает стремиться к бесконечности. Существуют критические значения как начальной концентрации кокса, так и начальной концентрации кислорода, при которых это явление может иметь место. Так как концентрация кокса, при которой должна начаться регенерация, обычно заранее задана, то начальная концентрация кислорода становится переменной, определяющей режим процесса.
Были сделаны попытки ограничить максимальное повышение температуры с помощью различных методов. Эффективность использования пара в регенераторе для ограничения повышения температуры оказалась сомнительной, в то время как разбавление слоя катализатора с использованием металлов может привести к необычным и даже противоположным эффектам, как будет показано ниже. Поскольку монооксид и диоксид углерода являются первичными продуктами окисления углерода, то попытки уменьшить вторичное окисление СО в СО2
важны для управления процессом повышения температуры.
Обычно выдается следующая рекомендация для исключения перегревов в слое [45]: выбрать небольшую начальную концентрацию кислорода и по мере формирования фронта горения постепенно ее повышать. С другой стороны, разогревы в слое сильно чувствительны при невысокой начальной концентрации кислорода к входной температуре. Поэтому при условии образования фронта горения выгодным является снижение температуры газа на входе в регенератор. Это позволяет уменьшить энергетические затраты на подогрев регенерационного газа и увеличить содержание в нем кислорода. Последнее обстоятельство позволит снизить расход регенерационного газа и уменьшить затраты на его циркуляцию.
1.8.1 Регенерация немодифицированных цеолитов
Наибольшее внимание исследователи уделяют вопросам регенерации немодифицированных цеолитов. Из предложенных вариантов в первую очередь необходимо отметить способ окислительной регенерации [40-42]. В работе [41] показано, что существенное влияние на скорость процесса регенерации оказывает температура процесса, содержание кислорода и водяного пара в регенерирующей смеси. Кислород не оказывает влияния на кинетику выгорания кокса. Окислительная регенерация полностью восстанавливает активность катализатора [42]. Удаление кокса происходит без разрушения кристаллического каркаса цеолита.
Методами температурно-программированного окисления в среде воздуха и кислорода (1% О2
в аргоне), масс- и ИК-спектроскопии изучена [40] регенерация закоксованных образцов цеолита ZSM-5 с различным содержанием кокса. Установлено, что определенная часть кокса (независимо от общего содержания кокса в образце) при температуре 300-450 0
С легко окисляется с образованием карбонильных соединений, которые при более высокой температуре окисляются до СО и СО2
. Количество образующегося СО2
коррелируется с содержанием кокса в образце. Температурный максимум десорбции СО2
увеличивается с 400 0
С для образцов с низким содержанием кокса, до 600 0
С для образцов с содержанием кокса 4 %, что указывает на большую устойчивость кокса в последних. Окисление кокса коррелируется, по данным ИК-спектроскопии, с потерей метильных групп и ароматических колец. Конечной стадией регенерации цеолита является процесс декарбонилирования – декарбоксилирования.
В работе [43] представлены данные по влиянию регенерации на селективность и стабильность высококремнистого цеолита типа ZSM-5 в процессе конверсии метанола. Регенерация кислородом воздуха при температуре 773 К полностью удаляет кокс из катализатора. Показано, что изменение селективности и стабильности под действием воды, выделившейся в конверсии метанола в углеводороды, приводит к обратимой дезактивации цеолита за счет перехода одних типов кислотных центров в другие и необратимой потере активации вследствие деалюминирования катализатора.
Авторами [44] изучена кинетика регенерации СВК-цеолита (Si/Al=35) – катализатора процесса конверсии метанола. Показано влияние температуры и содержания кислорода в смеси для регенерации на скорость выжига коксовых отложений с поверхности катализатора.
Рядом фирм запатентованы способы регенерации немодифицированных цеолитов типа пентасил. Информация о них приведена ниже.
Дезактивированный коксом или его предшественниками катализатор синтеза углеводородов из метанола по процессу фирмы «Байер АГ» (ФРГ) [40] регенерируют при температуре 300-450 0
С с газом, содержащим кислород и водяной пар (можно О2
и N2
или дымовые газы) при парциальном давлении пара 0,1-50% от общего давления газа. В процессе регенерации количество азота понижают, а расход воды поддерживают постоянным.
Для регенерации катализатора изомеризации ксилола (фирма «Мобил ойл», США) через слой закоксованного катализатора в замкнутой циркуляционной системе пропускают газовую смесь. Смесь включает О2
, N2
и водяной пар с парциальным давлением последнего 0,07-2,8 МПа, регенерация проводится при температуре 370-540 0
С и времени контакта 12-120 ч. Периодически подается свежая смесь с тем, чтобы концентрация О2
на входе в реактор не превышала 1 % мол., а парциальное давление водяного пара более 0,703 МПа. Исходная смесь имеет точку росы -26 0
С.
Запатентован [40] способ регенерации катализатора конверсии олефинов в бензин газовой смесью с содержанием кислорода 0,7 % в начале процесса и 7 % в конце. Регенерацию проводят при температуре 450 -500 0
С. По окончании систему продувают азотом. В ряде работ сопоставлены результаты регенерации высококремнистых цеолитных катализаторов обычной обработкой О2
при 450 0
С и смесью О3
+ О2
при 150 0
С. Обнаружено, что при низкотемпературной регенерации с использованием озона увеличивается срок службы катализатора. Обработка катализатора смесью О3
+ О2
позволяет существенно снизить воздействие на цеолит паров воды, образующихся при окислении углеродсодержащих продуктов, которые формируются на поверхности катализатора в ходе конверсии. В конечном счете это способствует сохранению структуры цеолита.
1.8.2 Регенерация цеолитов, модифицированных неблагородными
металлами
Катализаторы, содержащие высококремнистые цеолиты, модифицированные неблагородными металлами, подвергают в основном окислительной регенерации. Так, немецкие исследователи запатентовали способ регенерации отработанного цеолитного катализатора (типа ZSM-5, модуль  ≥ 20), содержащего металл и (или) соединение металла из групп IIIа (0,01-5 % La) и (или) IIIв (0,01-5 % Ga), желательно также элемент из группы IV в (0,001-5 % Cr) при общей концентрации металлов до 15 % от массы цеолита, а также SiO2
и (или) Al2
O3
и (или) глину в количестве 1-90 % от общей массы катализатора, который содержит La и Cr в атомном соотношении 100:1 – 1:1 и (или) Ga:Cr 100:1 – 1:1. Количество металлсодержащего цеолита в катализаторе 10-99. Регенерацию отработанного катализатора осуществляют выжиганием углеродистых отложений при температуре менее 550 0
С в токе кислорода, разбавленного азотом, и последующим прокаливанием в токе воздуха более 1 ч при 550 0
С. ≥ 20), содержащего металл и (или) соединение металла из групп IIIа (0,01-5 % La) и (или) IIIв (0,01-5 % Ga), желательно также элемент из группы IV в (0,001-5 % Cr) при общей концентрации металлов до 15 % от массы цеолита, а также SiO2
и (или) Al2
O3
и (или) глину в количестве 1-90 % от общей массы катализатора, который содержит La и Cr в атомном соотношении 100:1 – 1:1 и (или) Ga:Cr 100:1 – 1:1. Количество металлсодержащего цеолита в катализаторе 10-99. Регенерацию отработанного катализатора осуществляют выжиганием углеродистых отложений при температуре менее 550 0
С в токе кислорода, разбавленного азотом, и последующим прокаливанием в токе воздуха более 1 ч при 550 0
С.
Фирмой ЮОП (США) запатентован способ окислительной регенерации катализатора процесса дегидроциклодимеризации углеводородов С1
-С10
9ZSM-5, 0,1-5 % Ga, 30-70 % P-содержащего оксида алюминия) [40]. После дезактивации катализатор помещают в регенератор, где при атмосферном давлении, объемной скорости 4800 ч-1
и линейной скорости потока 0,5 м/с проводят регенерацию в течение 7 ч кислородсодержащим газом. В процессе регенерации температуру постепенно повышают с 490 до 540 0
С, а содержание кислорода в газе увеличивают с 1 до 20 % мол. Катализатор выдерживает 5 циклов регенерации, в которых активность его полностью восстанавливается.
Дезактивированный в ходе гидрогенизационных процессов катализатор [46] регенерируют путем выжига углеродсодержащих соединений в среде килородсодержащего газа, включающей в себя стадии десорбции углеводородов с поверхности катализатора, осуществляемой в реакторе технологической установки, и выжига продуктов их уплотнения на специализированной установке во вращающейся печи непрямого нагрева при температуре 450 - 5500
С и давлении 0,4-1 атмосферы.
Для регенерации отечественного катализатора ароматизации фракции С3
-С4
сотрудники НПО «Леннефтехим» также предложили [47] способ окислительной регенерации, которая сводится к воздушному выжиганию кокса с предварительной продувкой азотом или без нее в том же реакторе при температуре до 600 0
С. Полученные данные позволяют оптимизировать стадию регенерации цеолитсодержащего катализатора процесса «алифор».
Авторами [48] изучена кинетика регенерации СВК-цеолита (Si/Al=35, степень кристалличность 100 %) – катализатора процесса конверсии метанола. Показано влияние температуры и содержания кислорода в смеси для регенерации на скорость выжига коксовых отложений с поверхности катализатора.
Рядом фирм запатентованы способы регенерации немодифицированных цеолитов типа пентасил. Информация о них приведена ниже.
Дезактивированный коксом или его предшественниками катализатор синтеза углеводородов из метанола по процессу фирмы «Байер АГ» (ФРГ) [48] регенерируют при температуре 300-450 0
С с газом, содержащим кислород и водяной пар (можно О2
и N2
или дымовые газы) при парциальном давлении пара 0,1-50% от общего давления газа. В процессе регенерации количество азота понижают, а расход воды поддерживают постоянным.
Для регенерации катализатора изомеризации ксилола (фирма «Мобил ойл», США) [49] через слой закоксованного катализатора в замкнутой циркуляционной системе пропускают газовую смесь. Смесь включает О2
, N2
и водяной пар с парциальным давлением последнего 0,07-2,8 МПа, регенерация проводится при температуре 370-540 0
С и времени контакта 12-120 ч. Периодически подается свежая смесь с тем, чтобы концентрация О2
на входе в реактор не превышала 1 % мол., а парциальное давление водяного пара более 0,703 МПа. Исходная смесь имеет точку росы -26 0
С.
Дезактивированный в ходе гидрогенизационных процессов катализатор [50] регенерируют путем выжига углеродсодержащих соединений в среде килородсодержащего газа, включающей в себя стадии десорбции углеводородов с поверхности катализатора, осуществляемой в реакторе технологической установки, и выжига продуктов их уплотнения на специализированной установке во вращающейся печи непрямого нагрева при температуре 450 - 5500
С и давлении 0,4-1 атмосферы.
Недостатком процесса окислительной регенерации является снижение активности катализатора после попадания на него влаги, образующейся при выжигании углеродистых отложений. Фирмой ЮОП предложен способ устранения этого недостатка для оптимизации в промышленных условиях процесса ароматизации в реакторах и регенераторах с движущимся (сверху вниз) слоем мелкосферического катализатора. Выходящий из реактора катализатор (SiO2
:Al2
O3
≥12; 0,1-5 % Ga; P:Al (в Al2
O3
) 1:1-100), содержащий до 20 % кокса (обычно 5-7 %), подается в зону выжигания регенератора, куда противотоком поступает нагретый сухой воздух, возможно смешанный с осушенными газами рецикла. Ранее катализатор проходит зону осушки, где контактирует с нагретым сухим воздухом или с газами рецикла. После выхода из нижней части регенератора катализатор вновь подают в верхнюю часть реактора. Газы из зоны выжигания частично смешивают с воздухом и после осушки и подогрева вновь используют для выжигания кокса, часть их выводится в атмосферу. Осушители воздуха и газов рецикла заполняют цеолитом.
Описаны также примеры регенерации катализаторов с использованием сверхкритических сред. Так, цеолитный катализатор алкилирования бутиленов изобутаном, который обычно дезактивируется через 8-10 ч работы полностью регенерируется в сверхкритических условиях, причем последние могут быть реализованы непосредственно в процессе алкилирования (сверхкритические углеводороды). После 34 циклов реакция-регенерация активность составляет 90% от исходной, при этом значительно сокращается время регенерации, увеличивается срок жизни катализатора, и число рабочих циклов [51].
Разработанные катализаторы процесса Цеоформинга характеризуются низкой активностью в реакциях коксообразования, что позволяет проводить цикл реакции непрерывно в течение 220-300 ч, после чего проводится регенерация катализатора. Общий срок службы катализатора — более одного года.
При введении в состав катализатора до 0,3% мас. благородного металла цикл его непрерывной эксплуатации повышается до 800 ч [52].
1.9 Критерии оценки ущерба окружающей среде при производстве моторных топлив
В настоящее время санкции за нанесенный окружающей среде ущерб при работе любого предприятия являются нежелательным и весьма болезненным аргументом, свидетельствующим о необходимости проведения природоохранных мероприятий. В процессах глубокой переработки нефти, и в частности, при получении автомобильных топлив облагораживанием низкооктанового углеводородного сырья в различных каталитических процессах, имеются следующие потенциальные источники выбросов в окружающую среду:
1) газообразные, образующиеся:
а) на стадии облагораживания при утечке газообразный продуктов;
б) на стадии регенерации катализатора – при наличии в газах регенерации, сбрасываемых в атмосферу, помимо СО2
(считающегося относительно безопасным компонентом, если его количество не превышает определенных нормативов), других компонентов, являющихся токсинами (СО, окислы серы и азота);
в) на стадии использования полученного бензина – в виде выхлопных газов автомобиля;
2) жидкие – утечки сырья или продуктов через дефекты в аппаратуре, сточные и промывные воды;
3) твердые – чаще всего это отработанные катализаторы и адсорбенты.
Чем больше текущие затраты на природоохранную деятельность, тем меньше экономический ущерб, и наоборот. Увеличение текущих затрат не означает роста общих затрат на производство, так как на величину этого роста уменьшается экономический ущерб предприятия, который также включен в себестоимость выпуска товарной продукции.
Общие затраты на производство увеличатся лишь при превышении текущих затрат на природоохранную деятельность над издержками на охрану окружающей среды. Сокращение текущих затрат на природоохранную деятельность не означает снижения себестоимости продукции. Последнее достигается лишь при повышении эффективности природоохранных мероприятий.
Текущие затраты на природоохранную деятельность носят активный преобразующий, а не пассивный компенсирующий характер, т.е. текущие затраты направлены на устранение причины – загрязнение окружающей среды, а экономический ущерб является следствием этого загрязнения [53].
Отработка технологии на стадии лабораторных и пилотных испытаний должна помимо основных требований к процессу, обеспечивающих качественное и количественное получение целевых продуктов при оптимальных технологических параметров, должна обеспечивать еще и оценку возможного экологического ущерба от рекомендуемой технологии. Такое комплексное исследование технологических и экологических критериев позволит избежать последствий по внедрению мероприятий, нейтрализующих ущерб окружающей среде.
Требования экологического законодательства по уменьшению в топливах для автотранспорта содержания серы, ароматических углеводородов, бензола, при одновременном повышении их детонационной стойкости, вызывают серьезные изменения в технологии производства компонентов и балансах компаундирования автомобильных бензинов на НПЗ, что сопряжено со значительными капитальными затратами [3]. Большую проблему представляет обеспечение требований стандарта по содержанию бензола и ароматических углеводородов, так как основным высокооктановым компонентом автомобильных бензинов, вырабатываемых на большинстве предприятий России, является бензин каталитического риформинга, содержание бензола в котором доходит до 7% . Доля риформата в составе бензинового фонда России превышает 50%, а его содержание в высокооктановых бензинах достигает 90%. В нефтепереработке развитых стран мира бензин каталитического риформинга, как компонент товарных автомобильных бензинов, постепенно утрачивает свое былое значение. В последние годы активность по созданию на НПЗ мира новых установок каталитического крекинга приобрела рекордно высокий уровень. Производство бензинов класса Евро-4 и выше без каталитического крекинга и сопряженных с ним процессов (алкилирование, получение оксигенатов, полимеризация и др.) вообще невозможно. Для справки: доля каталитического крекинга относительно мощностей первичной переработки нефти в России составляет 6,6 %, в США – 35 % а в странах западной Европы – 15,4 %. Соответственно можно сделать вывод о необходимых масштабах модернизации отечественной нефтепереработки. И это не только колоссальные финансовые затраты, и время, которого практически нет. За 1,5 года, которые остались до перехода на производство топлив Евро-4, комплекс каталитического крекинга не построишь.
По данным департамента стратегического развития НК «Лукойл» переход российской промышленности к производству бензинов по стандарту Евро-5 потребуется 80 млрд. $. Объявленные инвестиционные проекты НПЗ до 2017 г. составляют $ 49 млрд. и не обеспечат в полной мере выполнение регламента по бензинам к 2010 г. [54].
Необходимо оценить потребность в топливах того или иного экологического класса с учетом структуры существующего и перспективного автопарка. Исходя из этого, определить оптимальный экономически целесообразный уровень требований к качеству топлив с учетом общепринятой мировой практики, которая основывается на том, что:
- первое это автомобиль;
- второе современному автомобилю соответствующее топливо.
В настоящее время в преддверии приближающейся олимпиады в Сочи 2014 года в Южном федеральном округе развернулась кампания по наращиванию мощностей действующих и строительству новых нефтеперерабатывающих предприятий. Основная цель это кампании – обеспечение моторным топливом техники (сначала – строительной, потом – авто- и авиа-). И в свете этого стремительно приближающегося события особенно актуальной является разработка технологий производства моторных топлив, отвечающих современным требованиям международных стандартов, и при этом наносящих минимальный ущерб курортной зоне Краснодарского края.
Таким образом, анализ информационных источников показал, что:
- сегодня проблема производства моторных топлив, отвечающих экологическим требованиям международных стандартов, является особенно актульной;
- для производства моторных топлив необходимо строительство установок глубокой переработки нефти с использованием каталитических процессов;
- среди катализаторов для получения моторных топлив наиболее перспективными являются цеолитсодержащие;
- состав используемого катализатора влияет не только на технологические параметры процесса, но и на его экологические характеристики.
Основной целью настоящей работы мы определили разработку и совершенствование эколого-безопасных способов регенерации цеолитсодержащих катализаторов, используемых при производстве высокооктановых моторных топлив.
В недавних исследованиях, проводимых в лабораториях КубГТУ, были определены наиболее оптимальные сочетания модифицирующих добавок для этих катализаторов [55]. Однако определение времени их стабильной работы, а также выявление влияния модифицирующих агентов на эту характеристику до сих не проводились. Поэтому в данной работе предстояло выполнить следующие задачи:
- Исследование влияния катализаторов облагораживания низкооктановых углеводородных фракций на природные экосистемы, поиск способов повышения их стабильности и селективности при производстве высокооктановых компонентов моторных топлив, отвечающих современным экологическим требованиям определение эксплуатационных свойств цеолитсодержащих катализаторов облагораживания прямогонных бензиновых фракций;
- выявление условий эксплуатации катализаторов, способствующих получению компонентов моторных топлив с пониженным содержанием токсичных ароматических соединений;
- исследование закономерностей коксообразования и подбор режимов восстановления свойств контактов, с минимальным содержанием токсичных компонентов в газах регенерации, отходящих с установки
|