МІНІСТЕРСТВО ОСВІТИ І НАУКИ УКРАЇНИ
ДОНЕЦЬКИЙ НАЦІОНАЛЬНИЙ УНІВЕРСИТЕТ
ХІМІЧНИЙ ФАКУЛЬТЕТ
МАГІСТЕРСЬКА РОБОТА
на тему: ВПЛИВ СТРУКТУРИ АЛІФАТИЧНИХ КАРБОНОВИХ КИСЛОТ ТА ТРЕТИННИХ АМІНІВ НА КАТАЛІТИЧНИЙ АЦИДОЛІЗ ЕПІХЛОРГІДРИНУ
Магістр: Козорезова Олена Ігорівна
Спеціальність: 8.070301 "Хімія"
Затверджена наказом № 125/08 від 04.02.2005 року
Керівник:к.х.н., доц. Швед О.М.
асистент Усачов В. В.
Донецьк — 2005
АННОТАЦИЯ
Козорезова Е. И. "Влияние структуры алифатических карбоновых кислот и третичных аминов на каталитический ацидолиз эпихлоргидрина".Работа на соискание магистра химии по специальности 8.070301 "Химия", Донецкий национальный университет. Донецк, 2005.
Изучены кинетические закономерности реакции каталитического ацидолиза эпихлоргидрина уксусной кислотой и ее производными в интервале температур 300
– 600
С при концентрации катализатора 0,00125 – 0,005 М. Установлен нулевой порядок реакции по кислотному реагенту и первый порядок реакции по катализатору. Выявлена низкая чувствительность реакции к основности катализатора. Расчитаные активационные параметры реакции при сопоставлении уравнений Аррениуса и Эйринга отвечают таковым для реакций бимолекулярного нуклеофильного замещения. Изучено влияние природы заместителя в уксусной кислоте на скорость ацидолиза эпихлоргидрина. Получена единая изикинетическая зависимость, указывающая на неизменность механизма реакции в данных условиях.
Ключевые слова:
кинетика, эпихлоргидрин, порядок реакции, энергетические параметры, уксусная кислота, производные уксусной кислоты, катализатор основной природы, структура кислотного реагента.
THE SUMMARY
Kozorezova H. I. "Influence of structure of aliphatic monobasic carboxylic acids and tertiary amines on catalytic acidolysis of epichlorohydrin".
Work on competition of chemical master on a speciality 8.070301 "Chemistry", Donetsk national university. Donetsk, 2005.
Kinetic laws of catalytic acidolysis of epichlorohydrin by an acetic acid and its derivatives in an interval of temperatures 300
– 600
С at concentration of the catalyst 0,00125 - 0,005 mole/l are investigated. The zero order of reaction on acid reagent and the first order of reaction on the catalyst are established. The low sensitivity of reaction to basis of the catalyst is revealed. The calculated activation parameters by confrontation of Arrhenius and Iring equations are identical to those for the reactions proceeding on the mechanism of dimolecular nucleophilic substitution. The influence of a nature of the assistant in an acetic acid on speed of acidolysis of epichlorohydrin is investigated. The uniform isokinetic dependence indicating an invariance of the mechanism reactions in the given conditions is received.
Key words:
kinetics, epichlorohydrin, order of reaction, power parameters, acetic acid, derivatives of an acetic acid, catalyst of the basic nature, structure of an acid reagent.
Зміст
ВСТУП
1.ЛІТЕРАТУРНИЙ ОГЛЯД
1.1 Загальні уявлення про розкриття епоксидного циклу під дією нуклеофільних реагентів
1.2 Напрямок розкриття кільця епокисей
1.3 Напрямок розкриття α-оксидного кільця в реакції епіхлоргідрину з карбоновими кислотами при основному каталізі
1.4 Вплив структури кислотного реагенту на швидкість реакції
1.5 Вплив концентрації каталізатору на швидкість реакції
1.6 Вплив структури каталізатору на швидкість реакції
1.7 Каталіз реакції фенілгліцидилового ефіру з карбоновими кислотами у присутності каталізатору N,N-диметиланіліну
1.8 Вплив температури на швидкість реакції карбонових кислот з епоксидними сполуками
2. ЕКСПЕРИМЕНТАЛЬНА ЧАСТИНА
2.1 Синтез та очистка речовин
2.1.1 Епіхлоргідрин
2.1.2 N,N-диметиланілін
2.1.3 4-Br-N,N-диметиланілін
2.1.4 3-NO2
-N,N-диметиланілін
2.1.5 γ-Піколін (4-метилпіридин)
2.1.6 Тетраетиламоній бромід
2.1.7 Триметилоцтова (півалева) кислота24 2.1.8. Етоксіоцтова кислота
2.1.9 Феноксіоцтова кислота
2.1.10 Феніл оцтова кислота
2.1.11 Масляна кислота
2.1.12 Ізомасляна кислота
2.1.13 Пропіонова кислота
2.2 Методика кінетичних вимірювань
2.3 Математична обробка експериментальних даних
2.4 Техніка безпеки
2.4.1 Робота з епіхлоргідрином
2.4.2 Робота з оцтовою кислотою
2.4.3 Робота з амінами
2.4.4 Робота з кислотами та лугами
3. РЕЗУЛЬТАТИ ТА ЇХ ОБГОВОРЕННЯ
3.1 Визначення порядку реакції за карбоновою кислотою
3.2 Визначення порядку реакції за каталізатором
3.3 Вплив структури кислотного реагенту на швидкість ацидолізу ЕХГ
3.4 Вплив температури на швидкість ацидолізу ЕХГ
ВИСНОВКИ
ЛІТЕРАТУРА
ВСТУП
Вже протягом декількох десятиліть не вгасає інтерес до реакції епіхлоргідрину з органічними кислотами [1-4] з метою отримання оксіхлорпропільних похідних карбонових кислот, які є важливими проміжними продуктами для отримання поверхнево-активних речовин, антистатиків, охолоджувачів, антикорозійних покриттів та епоксидних матеріалів з рядом цінних властивостей: термостійкість, хемостійкість, атмосферостійкість, тривалість до механічних дій і т. ін. [5] Окрім цього, вивчення закономірностей механізму та напрямку розкриття оксиранового циклу нуклеофільними реагентами уявляє собою актуальну задачу та є складовою частиною досліджень реакцій нуклеофільного заміщення:
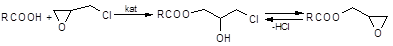 (1) (1)
Не дивлячись на велику кількість досліджень, що присвячені кінетиці та каталізу даної реакції, як і раніше залишається відкритим питання відносно механізму ацидолізу епіхлоргідрину, який проводиться в умовах, що є ідентичними до промислових (у середовищі 1-хлор-2,3-епоксіпропану). Більшість досліджень, як правило, виконано у розчинниках: хлорбензол [1], нітробензол [3], ДМФА [3], ДМСО [4], які можуть неоднозначно впливати на кінетику та механізм реакції (1) [4].
Актуальним є дослідження реакційної здатності карбонових кислот в реакції з α-оксидами для встановлення впливу їхньої структури на швидкість реакції.
Метою даної роботи є вивчення кінетичних закономірностей реакції оцтової кислоти та її похідних з епіхлоргідрином, встановлення впливу природи замісника в оцтовій кислоті, концентрації та структури каталізатору, а також температури на швидкість ацидолізу епіхлоргідрину, який виступає і в якості реагенту, і в якості розчинника. Вузловим моментом дослідження є встановлення порядку як реакції у цілому, так і за окремими компонентами.
В якості об’єкту дослідження була обрана реакція триметилоцтової, ізомасляної, масляної, пропіонової, оцтової, фенілоцтової, етоксіоцтової та феноксіоцтової кислот з епіхлоргідрином у присутності ряду каталізаторів – амонійових солей та амінів, а саме: (C2
H5
)4
NBr, 4-Me-C5
H4
N, C6
H5
N(CH3
)2
, 4-Br-C6
H4
N(CH3
)2
, 3-NO2
-C6
H4
N(CH3
)2
.
Робота складається з літературного огляду, експериментальної частини, де наведені методики синтезу та очистки речовин, кінетичних вимірів, математичної обробки результатів. Робота завершується результатами, їх обговоренням, висновками та списком літературних джерел.
1. ЛІТЕРАТУРНИЙ ОГЛЯД
1.1 Загальні уявлення про розкриття епоксидного циклу під дією нуклеофільних реагентів
Реакція α-оксидів з карбоновими кислотами у присутності основних каталізаторів вивчається як у надлишку α-оксиду, так і у надлишку кислоти [6]. З високим виходом при цьому утворюються відповідні оксіефіри. Однак питання про напрямок розкриття α-оксидного кільця є дискусійним.
Відомо, що реакції заміщених оксидів етилену з амінами та нуклеофільними реагентами типу спиртів та фенолів протікають у більшості випадків у відповідності з правилом Красуського [6]. Кислі реагенти, а також нейтральні у присутності кислих каталізаторів взаємодіють з α-оксидами всупереч цьому правилу, однак частіше при цьому утворюється суміш ізомерів – первинних та вторинних спиртів [3].
Літературні дані про напрямок розкриття α-оксидного кільця досить суперечливі. Це може бути пояснено недосконалістю методів розділення та аналізу двох ізомерних продуктів, труднощами отримання можливих ізомерів зустрічним синтезом, можливістю взаємоперетворення ізомерних продуктів за рахунок внутрішньомолекулярної міграції ацильних радикалів [6].
1.2 Напрямок розкриття α-окисного циклу
В епоксидному циклі є два атоми карбону та будь-який з них може підвергатися нуклеофільній атаці. У симетричному епоксидному циклі, наприклад, оксиді етилену, обидва атоми карбону є еквівалентними і атака направляється по одному та другому атомам випадковим чином. Але у несиметричному епоксидні атоми карбону не є еквівалентними та будова продукту реакції, що утворюється, визначається тим, який з цих атомів атакується переважно [4].
Доводиться, що переважне місце атаки залежить головним чином від того, чим каналізується реакція – кислотами чи основами:
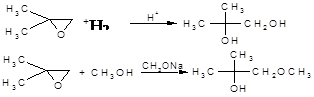
В цих реакціях, як і в загалі завжди, нуклеофіл атакує більш заміщений атом вуглецю при розщепленні, що каналізується кислотами, та менш заміщений атом карбону при розщепленні, що каналізується основами [4].
Це вказує на реалізацію двох механізмів – SN1
та SN2
. Однак є дані, які чітко показують, що обидва механізми відносяться до SN2
– типу: розщеплення зв’язку С─О та атака нуклеофіла протікають в одну стадію.
В реакції SN2
атом карбону віддає електрони групі, що відходить, та отримує електрони від нуклеофіла, в результаті чого він не отримує помітного позитивного чи негативного заряду у перехідному стані; електронні фактори не суттєві для хода реакції, і вона контролюється просторовими факторами. Однак у розщепленні епокисі, що каналізується кислотою, зв’язок С─О, вже послаблений кутовою напругою у трьохчленному кільці, ще більш послаблюється внаслідок протонування. Хоча у перехідному стані спостерігається як розрив, так і утворення зв’язку, розрив зв’язку протікає у більшому ступені, ніж його утворення; група, що відходить, відтягує за собою електрони у значно більшому ступені, ніж нуклеофіл надає їх, і атом карбону набуває значного позитивного заряду [4].
У цьому випадку просторові перешкоди значно менш важливі, оскільки як група, що відходить, так і нуклеофіл знаходяться достатньо далеко. Стійкість перехідного стану визначається в основному електронними, а не просторовими факторами: атакується не найменш заміщений атом карбону, а атом карбону, який найкраще може розмістити позитивний заряд (про таку реакцію говорять, що вона має значний SN1
– характер).
SN2
– розкриття кільця, що каналізується кислотою:

У розщепленні, що каталізується основами, група, яка відходить, значно менш ефективна, а нуклеофіл є дуже ефективним. Розрив та утворення зв’язку відбуваються майже в однаковій мірі, і реакційна здатність контролюється, як завжди, просторовими факторами.
SN2
– розкриття кільця, що каналізується основою:
Нуклеофільна атака епоксидного циклу у некислому середовищі була добре вивчена; широкий діапазон реакцій вказує на другий порядок реакції, що відповідає SN2
– механізму [7].
Пакер та Ісакс висказали ідею, що розрив зв’язку є більш важливим фактором, ніж його поява в утворенні перехідного стану [4].
Механізм реакції розкриття епоксидного циклу у кислому середовищі не був вивчений досконально.
Існує майже рівна імовірність на енергетичному рівні двох механізмів. Таким чином, кінетичний критерій щодо механізму є незадовільне ним та можуть бути використані альтернативні критерії [7].
Прітчард та Лонг вивчали кінетику та співвідношення отриманих продуктів реакції гідролізу алкілзаміщених оксидів етилену у кислому середовищі [7]. Виходячи з кореляції між константами швидкості та функції Гамету Н0
, вони зробили висновок, що реакція протікає за механізмом А1
.
Другим критерієм щодо встановлення механізму розкриття епоксидного циклу було використання рівняння Тафта:
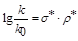 (1.1), де (1.1), де
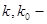 константи швидкості заміщеного епоксидну та пропілен оксиду відповідно; константи швидкості заміщеного епоксидну та пропілен оксиду відповідно;
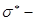 константа замісника; константа замісника;
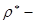 константа реакції константа реакції
Вчені отримали величину 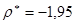 та висловили думку, що це є вказівкою на протікання реакції за механізмом А1
[7]. та висловили думку, що це є вказівкою на протікання реакції за механізмом А1
[7].
Однак Пакер та Ісакс помітили, що від’ємне значення  не є критерієм А1
– механізму за тією причини, що від’ємне значення вказує на зростання швидкості реакції у випадку донорних замісників, які зменшують електронну густину на атомі оксигену епоксидну, збільшення кількості епоксидну призводить до вихідної рівноваги і, таким чином, збільшення швидкості реакції спостерігається в обох випадках: як при А1
-, так і при А2
– механізмі [7]. не є критерієм А1
– механізму за тією причини, що від’ємне значення вказує на зростання швидкості реакції у випадку донорних замісників, які зменшують електронну густину на атомі оксигену епоксидну, збільшення кількості епоксидну призводить до вихідної рівноваги і, таким чином, збільшення швидкості реакції спостерігається в обох випадках: як при А1
-, так і при А2
– механізмі [7].
Третій критерій заснований на гіпотезі, що механізм (А1
чи А2
) залежить від замісника. Відношення констант швидкості другого порядку реакції гідролізу епоксидів в оксиді дейтерія та у воді, отримані Прітчардом та Лонгом, були в інтервалі 1,9─2,2; це, на їхню думку, вказувало на А1
– механізм. У більш детальному аналізі Свейн та Торнтон довели, що ці відомості вказують на механізм А2
.
У четвертий критерій покладено важливість значення ентропії активації ΔS#
реакцій, що вивчають.
Таблиця 1.1 - Значення ентропії активації та механізм для реакцій карбонових кислот, естерів та третбутилгалогенідів із спиртами як розчинником [7]
| Субстрат |
Розчинник |
Т, 0
С |
Механізм |
ΔS#
, кал/моль•К |
| 1 |
2 |
3 |
4 |
5 |
Ph-COOH
o-CH3
C6
H4
COOH
o-ClC6
H4
COOH
CH3
COOH
|
CH3
OH
CH3
OH
CH3
OH
C2
H5
OH
|
40
40
40
25
|
A2
A2
A2
A2
|
-24.00
-29.60
-27.90
-35.70
|
CCl3
COOH
p-CH3
OC6
H4
COOCH3
C6
H5
COOCH3
p-NO2
C6
H4
COOCH3
(CH3
)3
CBr
(CH3
)3
CCl
(CH3
)3
CJ
(CH3
)3
CCl
(CH3
)3
CCl
|
C2
H5
OH
CH3
OH-H2
O (6:4)
CH3
OH-H2
O (6:4)
CH3
OH-H2
O (6:4)
C2
H5
OH-H2
O (4:1)
C2
H5
OH-H2
O (4:1)
C2
H5
OH-H2
O (4:1)
CH3
OH
C2
H5
OH
|
25
30
30
30
25
25
25
25
25
|
A2
A2
A2
A2
A1
A1
A1
A1
A1
|
-33.60
-29.80
-30.50
-30.70
+0.27
-7.06
+0.79
-4.51
-5.50
|
З табл. 1.1 видно, що величина ентропії активації ΔS#
для реакцій, що йдуть за механізмом А2
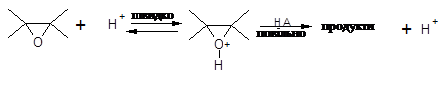
менша, ніж для реакцій механізму А1
:
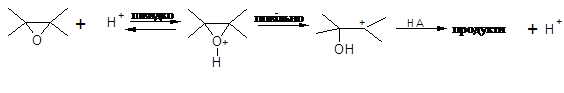
Прітчард та Лонг [7] вивчали реакції з моно- та дизаміщеними оксидами етилену. Вони виявили, що залежність у координатах lg k─σ*
(де k – константа швидкості першого порядку; σ*
- параметр Тафта) має нелінійний характер у випадку 1,1 – дизаміщених епоксидів. До цього моменту вони інтерпритували свої результати як протікання гідролізу за механізмом А1
у всіх випадках. Отже, Прітчард та Лонг прийшли висновку, що 1,1 – дизаміщені епоксиди є відхиленням від механізму А1
[7].
Найбільш вивченими є реакції оксиду пропілену, при взаємодії якого з кислотами утворюється суміш ізомерів. Кількість продукту аномального приєднання складає від 20 до 50% у залежності від природи кислоти (були вивчені оцтова, ди-, та три хлороцтова, акрилова та метакрилова кислоти), наявності каталізатора та його природи, а також температури [6].
При взаємодії з кислотами оксиду стиролу утворюється переважно аномальний продукт, причому можлива його подальша ізомеризація у нормальний [6].
У результаті реакції фенілгліцидилового ефіру з капроновою кислотою у присутності гідроксидів калію, натрію, ацетату натрію, тригептиламіну та без каталізатору утворюється 3 - 5% продукту аномального приєднання [6].
Дані про продукти взаємодії карбонових кислот з епіхлоргідрином надто обмежені. Так Бігот вказував, що в результаті реакції еквівалентних кількостей епіхлоргідрину та оцтової кислоти при 1800
С в основному утворюється 1,2 – хлоргідриновий ефір, але є і деяка кількість 1,3 – ізомеру, а в реакції епіхлоргідрину з акриловою та метакриловою кислотами аномального продукту не спостерігалося [6].
1.3 Напрямок розкриття α-оксидного кільця в реакції епіхлоргідрину з карбоновими кислотами при основному каталізі
Для дослідження напрямку розкриття α-оксидного кільця епіхлоргідрину масляною, півалевою (триметилоцтовою) та бензойною кислотами у присутності ряду основних каталізаторів були використані методи газо-рідинної хроматографії (ГРХ) та протонного магнітного резонансу (ПМР). Реакцію проводили у двократному надлишку епіхлоргідрину при температурі 80 – 1100
С. У якості каталізаторів використовували триетиламін, тетраетиламоній йодид, гідроксид, хлорид та гідроортофосфат натрію у кількості 0,05 моль на 1 моль кислоти [6].
Методами елементного та функційного аналізу, а також ІЧ спектроскопії встановлено, що основним продуктом реакції (вихід 80 - 97%) є хлоргідриновий ефір карбонової кислоти [6].
Продукти реакції були досліджені методом ГРХ.
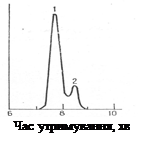
Рис. 1.1 Хроматограма суміші хлоргідринових ефірів масляної кислоти: 1 - 1,2-хлоргідриновий ефір; 2 - 1,3-хлоргідриновий ефір [6]
З рис. 1.1 видно, що хлоргідриновому ефіру відповідають два неповністю розділених піки, тобто у його складі наявні два ізомери, які неможливо розділити перегонкою [6].
Для підтвердження утворення аномального ізомеру було використано спектроскопію ПМР.
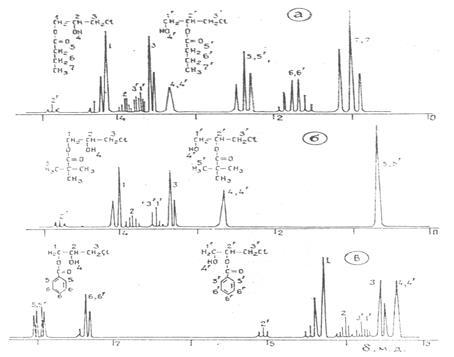
Рис. 1.2 ПМР спектри хлоргідринових ефірів масляної (а), півалевої (б), та бензойної (в) кислот [6]
Наявність резонансу метинового протону в області 5,2 – 4,9 м.д., обумовленого складноефірною групою, свідчить про присутність 1,3 – хлоргіринового ефіру. Кількість ізомеру, яка визначена методами ГРХ та ПМР, співпадає та складає 15 - 20% [6].
У табл. 1.2 наведені дані про ізомерний склад хлоргідринових ефірів, які отримані у різних умовах.
Таблиця 1.2 - Ізомерний склад хлоргідринових ефірів карбонових кислот в реакції ЕХГ з масляною, півалевою та бензойною кислотами в присутності каталізаторів основної природи [6]
| Каталізатор |
t, 0
С |
Кислота |
Вміст 1,3-ХГЕ, %
(за даними ГРХ)
|
NaOH
(C2
H5
)4
NJ
(C2
H5
)3
N
Na2
HPO4
NaCl
NaOH
NaOH
NaOH
(C2
H5
)4
NJ
(C2
H5
)4
NJ
(C2
H5
)4
NJ
(C2
H5
)4
NJ
|
80
80
80
80
80
80
90
100
80
90
100
110
|
CH3
CH2
CH2
COOH
CH3
CH2
CH2
COOH
CH3
CH2
CH2
COOH
CH3
CH2
CH2
COOH
CH3
CH2
CH2
COOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
(CH3
)3
CCOOH
|
21.9
18.1
17.1
15.1
14.7
15.3
17.5
18.3
18.0
17.8
18.0
18.0
|
З табл. 1.2 випливає, що зміна умов проведення реакції (природа каталізатора та температура) мало впливають на ізомерний склад продуктів взаємодії епіхлоргідрину з карбоновими кислотами [6].
1.4 Вплив структури кислотного реагенту на швидкість реакції
Для вивчення впливу структури кислотного реагенту на швидкість реакції було досліджено кінетику реакції оцтової, акрилової та метакриловлї кислот з 1,2-епоксі-3-феноксіпропаном у присутності ацетату хрому (ІІІ) як каталізатора при температурі 600
С [8]. Результати наведені в табл. 1.3.
Таблиця 1.3 - Константи швидкості реакції деяких карбонових кислот з 1,2-епоксі-3-феноксіпропаном у присутності ацетату хрому (ІІІ) [8]
| Кислота |
Т, 0
С |
k2
·102
, л/моль·с |
Оцтова
Акрилова
Метакрилова
|
60
60
60
|
0.552±0.030
1.075±0.055
1.068±0.022
|
З наведених величин констант швидкості таблиці 1.3 видно, що найшвидше з 1,2-епоксі-3-феноксіпропаном реагує акрилова кислота за даних умов проведення експерименту у той час як оцтова кислота – найповільніше [8].
У реакції перелічених вище кислот з епіхлоргідрином спостерігалася аналогічна залежність: швидкість реакції збільшувалася у ряду:
оцтова кислота < метакрилова кислота < акрилова кислота,
причому константи швидкості реакції з акриловою та метакриловою кислотами мало відрізнялися як і у випадку реакції цих кислот з 1,2-епоксі-3-феноксіпропаном [8].
Це пояснюють стеричним ефектом наведених кислот: найбільш об’ємна молекула метакрилової кислоти реагує важче з більш заміщеним атомом карбону 1,2-епоксі-3-феноксіпропана, ніж менш об’ємна молекула акрилової кислоти, не дивлячись на більш високу загальну швидкість взаємодії останнього [8].
Виходячи з експериментально отриманих величин констант швидкостей даних карбонових кислот, були розраховані величини  реакції карбонових кислот з 1,2-епоксі-3-феноксіпропаном, де реакції карбонових кислот з 1,2-епоксі-3-феноксіпропаном, де 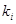 - константи швидкості акрилової та метакрилової кислот; - константи швидкості акрилової та метакрилової кислот; 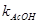 - константа швидкості оцтової кислоти. Ці величини набули значення 1.95 та 1.93 для акрилової та метакрилової кислот відповідно. Отже, за цими розрахунками встановлено, що при 600
С ненасичені кислоти мають подібну реакційну здатність [8]. - константа швидкості оцтової кислоти. Ці величини набули значення 1.95 та 1.93 для акрилової та метакрилової кислот відповідно. Отже, за цими розрахунками встановлено, що при 600
С ненасичені кислоти мають подібну реакційну здатність [8].
1.5 Вплив концентрації каталізатору на швидкість реакції
Для вивчення впливу концентрації каталізатору на швидкість реакції були досліджені кінетичні закономірності реакції оксиетилювання оцтової кислоти при каталізі ацетатами Li+
, Na+
, K+
, Cr3+
, [(Me)4
N]+
, [(Et)4
N]+
, [(Bu)4
N]+
. Експерименти були проведені при різних концентраціях солей, що вивчали при 900
С [9] (табл. 1.4).
Таблиця 1.4 - Залежність спостерігаємої константи швидкості від концентрації каталізатора [9]
| Скат
, моль/л |
САсОН
,
моль/л
|
kсп
·104
, с-1
|
Скат
, моль/л |
САсОН
,
моль/л
|
kсп
·104
, с-1
|
| CH3
COOLi |
(CH3
COO)3
Cr |
0.0980
0.1580
0.1920
0.2530
0.3546
|
16.02
16.00
15.90
15.88
15.82
|
3.28
4.27
5.33
6.48
8.73
|
0.0010
0.00295
0.00548
0.00842
0.0100
|
16.20
16.20
16.10
16.10
16.00
|
2.99
5.51
7.28
9.59
10.67
|
| CH3
COONa |
(Me)4
N+
CH3
COO–
|
0.0530
0.0840
0.1140
0.1550
0.1940
|
16.10
16.16
16.08
15.96
16.00
|
2.80
3.27
4.30
5.37
6.83
|
0.0710
0.0918
0.1508
0.2016
|
15.83
15.65
15.56
15.44
|
6.13
8.33
12.40
15.67
|
| CH3
COOK |
(Et)4
N+
CH3
COO–
|
0.1000
0.1450
0.2030
0.2480
|
15.88
15.68
15.80
15.78
|
4.58
5.95
7.57
9.80
|
0.0138
0.0750
0.1235
|
15.84
15.81
15.83
|
2.18
7.29
11.38
|
| (Bu)4
N+
CH3
COO–
|
0.0120
0.0564
0.0998
|
15.81
15.79
15.78
|
2.27
6.83
11.30
|
Як видно з експериментальних даних, досліджувані солі прискорюють реакцію прямопропорційно їх концентрації у реакційній масі [9].
При екстраполюванні експериментальних значень спостережених констант швидкостей до нульової концентрації каталізатору для всіх досліджуваних солей одержано одне і те ж значення, яке відповідає константі швидкості некаталітичної реакції, що узгоджується з даними [9] про паралельне протікання двох реакцій оксиду етилену – каталітичної (з участю солі) та некаталітичної [9].
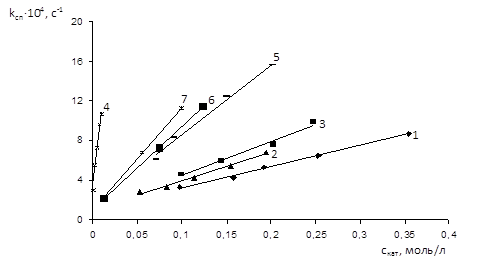
Рис. 1.3 Залежність спостереженої константи швидкості від концентрації каталізатора при 900
С: 1 - CH3
COOLi; 2 - CH3
COONa; 3 - CH3
COOK; 4 - (CH3
COO)3
Cr; 5 - (Me)4
N+
CH3
COO–
; 6 - (Et)4
N+
CH3
COO–
[9]
Отримані залежності описуються рівнянням:
kсп
= kн
+ kкат
·скат
(1.2), де
kн
– константа швидкості некаталітичної реакції, с-1
; kкат
- константа швидкості каталітичної реакції, л/(моль·с) [9].
Для встановлення порядку реакції за каталізатором було досліджено кінетику реакції оцтової, акрилової та метакрилової кислот з 1,2-епоксі-3-феноксіпропаном, яка каталізується ацетатом хрому (ІІІ) при 700
С. Концентрація каталізатору варіювалася у межах від 0.002 до 0.008 моль/л (у випадку акрилової та метакрилової кислот) та від 0.008 до 0.015 моль/л (у випадку оцтової кислоти) [8].

Рис. 1.4 Залежність ефективної константи швидкості карбонової кислоти від концентрації каталізатору при Т=700
С: 1 – CH2
=CH─COOH; 2 – CH2
=C(CH3
)─COOH; 3 – CH3
COOH [8]
Наведена вище графічна залежність у всіх випадках носить прямолінійний характер, що свідчить про перший порядок реакції за каталізатором [8].
1.6 Вплив структури каталізатору на швидкість реакції
Для вивчення впливу структури каталізатору була вивчена реакція оцтової, акрилової та метакрилової кислот з 1,2-епоксі-3-феноксіпропаном у присутності ряду метилзаміщених похідних піридину [8]. Збільшення швидкості реакції спостерігалося, коли піридин був заміщений метильним радикалом у третьому або четвертому положеннях, у той самий час, коли α-піколін (2-метилпіридин) виявився менш активним каталізатором, ніж незаміщений піридин. Подібна залежність каталітичної активності від структури каталізатору була виявлена для метилпохідних хіноліну [8].
При вивченні кінетичних закономірностей цієї реакції у присутності третинних амінів була виявлена залежність каталітичної активності від розміру катіона активної форми каталізатора. На основі отриманих експериментальних даних було встановлено, що із збільшенням радіусу катіона електростатична взаємодія між іонами ставала слабшою, нуклеофільність карбоксилат-аніону збільшувалася і збільшувалася каталітична активність. Такий взаємозв’язок був отриманий для реакції карбонових кислот з 1,2-епоксіпропаном та 1-хлор-2,3-епоксіпропаном. Однак, стеричні фактори можуть змінювати таку закономірність. Зменшення швидкості реакції спостерігалося для катіонів "великого розміру" у випадку 2-метилпіридину та 2-метилхіноліну [8].
1.7 Каталіз реакції фенілгліцидилового ефіру з карбоновими кислотами у присутності каталізатору N,N-диметиланіліну
Для пояснення прискорюючої дії жирноароматичних третинних амінів були запропоновані механізми загального основного та нуклеофільного каталізів реакції фенілгліцидилового ефіру з оцтовою та бензойною кислотами у присутності каталізатору N,N-диметиланіліну (ДМА), де амін виступає у ролі основи (реакція 1.1) або у ролі нуклеофіла (реакція 1.2) [9]:
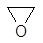 R3
N + R’
COOH R3
N + R’
COOH 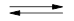 R3
N R3
N HOOCR’ HOOCR’
 [R3
NH]+
R’
COO─
(1.1) [R3
NH]+
R’
COO─
(1.1)
R3
N + R’
COOH + R’’
─  [R3
N─CH2
─CH2
R’’
(OH)]+
R’
COO─
(1.2) [R3
N─CH2
─CH2
R’’
(OH)]+
R’
COO─
(1.2)
Як було встановлено, каталітична активність жирноароматичних та аліфатичних третинних амінів дуже близька. Однак реалізація загального основного механізму каталіза жирноароматичними амінами (реакція 1.1) неможлива через їхню малу спроможність до протеолітичної взаємодії з карбоновими кислотами. Малоймовірна і реакція нуклеофільного механізму каталіза (реакція 1.2) через дезактивації неподільної пари електронів атома азота рπ-супрядженням. Останнє підтверджується даними про продукти реакції (табл. 1.5).
Таблиця 1.5 - Елементний склад хлоридів, виділених із реакційної суміші, а також речовин порівняння [9]
| Склад вихідної реакційної суміші, моль/л |
Елементний склад продуктів, % |
| ФГЕ |
СН3
СООН |
ДМА |
С |
Н |
N |
Cl |
3,0
0,8
|
2,0
4,5
|
0,7
0,12
|
63,0
59,9
|
6,93
7,45
|
5,92
6,79
|
15,4
18,7
|
| Речовини порівняння (розраховано): |
[(CH3
)2
C6
H5
NCH2
─CH(OH)─CH2
─OC6
H5
]Cl
(CH3
)2
C6
H5
N·HCl
|
66,2
60,7
|
7,46
7,69
|
4,55
8,85
|
11,5
22,4
|
У той же час карбоксилат-аніони можуть утворюватися при взаємодії аміна та кислоти через утворення комплексу з переносом заряду (КПЗ), при розпаді якого утворюються радикали та іони (реакція 1.3):
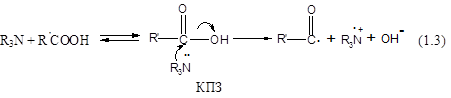
Для підтвердження утворення радикальних сполук, які свідчать про появу КПЗ (реакція 1.3), у реакційну суміш ФГЕ – С6
Н5
СООН – ДМА був введений метилметакрилат [9]. В умовах досліду був отриманий полімер, що відповідав структурі поліметилметакрилату. "Холостий" дослід показав, що у використаних умовах при відсутності кислоти ДМА полімеризацію метилметакрилату не викликає.
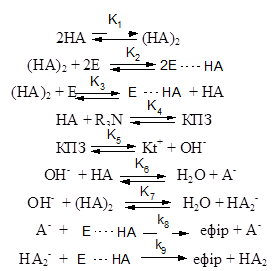
Отримані дані дозволили запропонувати схему протікання реакції α-оксиду з кислотою через стадію утворення КПЗ з урахуванням асоціативних взаємодій різного виду (НА – карбонова кислота, Е – епоксидна сполука):
1.8 Вплив температури на швидкість реакції карбонових кислот з епоксидними сполуками.
З метою вивчення впливу температури на швидкість реакції було проведено три серії досліджень реакції оксиду етилену з оцтовою та монохлороцтовою кислотами у кислому середовищі при температурах 900
С, 1000
С та 1050
С [3]. Дослідження проводилися у манометричному приладі у середовищі оцтової кислоти; концентрація оксиду етилену становила 0,49 – 0,52 М (при 900
С та 1000
С) та 0,37 – 0,38 М (при 1050
С). Отримані результати наведені у табл. 1.6:
Таблиця 1.6 - Константи швидкості та енергії активації реакції оксиду етилену з оцтовою кислотою [3]
| Реакція |
Константи швидкості, k·104
|
Еа
,
кДж/моль
|
| 900
С |
1000
С |
1050
С |
| При відсутності CH3
COONa (k, c-1
) |
2,00 |
4,10 |
6,02 |
80,7 |
| За участю CH3
COONa (k, л/моль·с) |
37,20 |
73,10 |
97,00 |
69,0 |
З наведених значень констант швидкості видно, що при збільшенні температури реакції від 900
С до 1050
С швидкість досліджуваної реакції збільшується у 2,05 рази при збільшенні температури на кожні 100
С.
При дослідженні впливу температури на швидкість реакції оцтової кислоти з оксидом етилену при каталізі ацетатом хрому (III) (CH3
COO)3
Cr [9] було проведено дві серії дослідів:
1. при концентрації каталізатору 0,001 М
2. при концентрації каталізатору 0,01 М
Отримані дані представлені у табл. 1.7
Таблиця 1.7 - Залежність константи швидкості реакції оксиетилювання оцтової кислоти від температури при каталізі ацетатом хрому (III) [9]
| Температура реакції, 0
С |
kеф.
·103
, c-1
|
| с0
оксиду етилену
= 1,0 М; скат.
= 0,001 М |
80
90
95
100
|
1,62±0,03
2,99±0,01
4,48±0,01
5,62±0,01
|
| с0
оксиду етилену
= 1,0 М; скат.
= 0,01 М |
80
90
100
|
6,89±0,01
11,30±0,01
17,80±0,01
|
Дослідження впливу температури на каталітичну та некаталітичну реакції МТГФК з епіхлоргідрином у присутності галогенідів тетраалкіламонія як каталізаторів [2] (табл. 1.8) показали помітний ріст констант швидкості реакції з підвищенням температури (γ=1,82).
Таблиця 1.8 - Константи швидкості реакції МТГФК з ЕХГ у присутності R4
NHal [2]
| Некаталітична реакція |
| Температура реакції, 0
С |
kеф.
·106
, c-1
|
90
100
110
|
2,56±0,01
6,61±0,01
19,70±0,01
|
| Каталітична реакція |
| Температура реакції, 0
С |
kеф.
·104
, л/моль·с |
70
80
85
90
|
1,51±0,01
2,78±0,01
3,81±0,01
5,78±0,01
|
Отже, на основі проведеного аналізу щодо впливу температури на швидкість реакції можна зробити висновок, що збільшення температури призводить до росту швидкості реакції.
2. Експериментальна частина
2.1 Синтез та очистка речовин
2.1.1 Епіхлоргідрин
Відпрацьований епіхлоргідрин містить домішки кислот і каталізаторів. З урахуванням того, що у деяких каталізаторів температура кипіння приблизно дорівнює температурі кипіння епіхлоргідрину, його промивають 15%-им розчином соляної кислоти (100 мл розчину на 1 л епіхлоргідрину), потім – декілька разів дистильованою водою і насиченим розчином KBr. Сушать над сульфатом натрію протягом доби. Після цього за допомогою азеотропної перегонки позбавляються залишків води. Отриманий безводний епіхлоргідрин переганяють при атмосферному тиску, відбираючи фракцію з температурою кипіння 116,5 – 1170
С (літ. Ткип.
= 1170
С) та nD
=1,4375 (літ. nD
= 1,5007); зберігають над молекулярними ситами [10,11].
2.1.2 N,N-диметиланілін
N,N-диметиланілін (ДМА) С6
Н5
N(СН3
)2
, що випускається у промисловості, кип’ятять з оцтовим ангідридом (на 100 мл диметиланіліна беруть 20 мл оцтового ангідриду) протягом 3 годин і переганяють [11]. Сушать протягом доби над NaOH (на 100 мл аміну беруть 10 г гранульованого гідроксида натрію) і переганяють, збираючи фракцію з Ткип.
= 192°С, nD
= 1,5578 (літ. Ткип.
= 193-194°С, nD
= 1,5581) [10,12].
2.1.3 4-Br-N,N-диметиланілін
Синтез виконують за схемою:
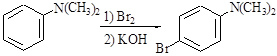
В трьохгорлу колбу об’ємом 500 мл, яка оснащена мішалкою, капельною воронкою и термометром (реакційна колба не повинна бути герметично закритою) поміщують розчин 12,1г (0,10моль) N,N-диметиланіліну в 30 мл діоксану і 5,6г (0,10 моль) КОН в 30 мл води. При інтенсивному перемішуванні додають з капельної воронки розчин 16,0г (0,10 моль) (5,1 мл) брому в 160 мл діоксану, слідкуючи за тим, щоб температура реакційної суміші була приблизно 50
С. Взагалі додавання брому займає близько двох годин.
Органічний шар відокремлюють і промивають 15 мл 40% розчину КОН. Потім відгоняють у вакуумі діоксан і залишок кристалізують з 70% етилового спирту ( на 1 г речовини беруть 3 мл розчинники). Ттопл.
= 54-55°С (літ. Ттопл.
= 54,5°С [10]).
2.1.4 3-NO2
-N,N-диметиланілін
3-нитро-N,N-диметиланілін очищують кристалізацією з петролейного ефіру ( на 1 г беруть 10 мл розчинника). Ттопл.
= 59-60°С (літ. Ттопл.
= 60°С [10]).
2.1.5 γ-Піколін (4-метилпіридин)
γ-Піколін (4-метилпіридин) очищують дворазовою перегонкою при зниженому тиску у присутності цинкового пилу: Ткип.
= 145°С, nD
= 1,2850 (літ. Ткип.
= 145,4°С, nD
= 1,2850 [10]).
2.1.6 Тетраетиламонійбромід
Тетраетиламонійбромід (C2
H5
)4
NBr ("ч") кристалізують із суміші бензол(б/в):этанол(б/в) у співвідношенні 3:2 (на 1 г речовини беруть 5 мл розчинника). Ттопл.
= 298-299°С, плавиться з розкладанням (літ. Ттопл.
= 299°С, розкладається [13,14].
2.1.7 Триметилоцтова (півалева) кислота
Синтез триметилоцтової кислоти здійснюють галоформним розщепленням пінаколіну [15]:
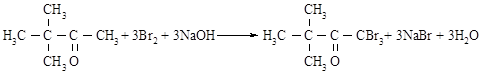
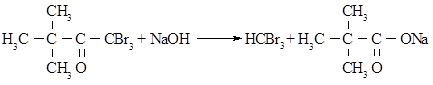
У двохгорлу колбу об’ємом 1 л, що обладнана мішалкою, вливають 630 мл розчину гідроксиду натрію (66,9 г NaOH у 568,0 мл дистильованої води), охолодженого до 0 ─ -1°С та поміщають у крижану баню. До розчину, що енергійно перемішується, через бічне горло доливають із крапельної воронки 97,4 г брому протягом 30-40 хвилин (температура повинна підтримуватися в межах -5 ─ +5°С). Потім у реакційну суміш при постійному перемішуванні з крапельної воронки доливають 20,3 г пінаколіна протягом 20-30 хвилин (температура повинна підтримуватися в межах -5 ─ +5°С). Після того, як долили весь пінаколін, розчин продовжують охолоджувати і перемішувати протягом 1 години. Потім колбу прикривають азбестовою ковдрою і продовжують перемішувати при кімнатній температурі протягом 5-6 годин до повного знебарвлення.
Потім колбу поміщають на водяну баню, закривають зворотним холодильником і нагрівають протягом 10-20 хвилин. Зворотний холодильник заміняють на прямий і відганяють з реакційної суміші бромоформ (Ткип
= 78°С), що відганяється разом з парами води. Коли у прямому холодильнику зникнуть маслянисті краплі, перегонку припиняють і реакційну суміш охолоджують до 50°С, після чого доливають 81,2 мл концентрованої соляної кислоти HCl. За рахунок теплоти нейтралізації, що виділяється, рідина закипає і частина триметилоцтової кислоти відганяється разом з водою. Коли мимовільне кипіння припиниться (~ 97°С), колбу нагрівають на водяній бані доти, поки не почне переганятися рідина, яка є важчою за воду. Тоді перегонку припиняють, а кислоту, яка відігналася, відокремлюють від води в ділильній воронці. Відділений водяний шар екстрагують двома порціями диетилового ефіру по 40 мл. Ефірні витяжки поєднують і сушать протягом доби над CaCl2
. Потім ефір відганяють на водяній бані, а триметилоцтову кислоту, що залишилася в колбі поєднують з відділеної від водяного шару. Для остаточного позбавлення від води, до отриманої кислоти доливають 30 мл зневодненого бензолу і воду відганяють за допомогою насадки Дина-Старка. Потім бензол відганяють при атмосферному тиску. Отриману зневоднену триметилоцтову кислоту переганяють двічі при зниженому тиску, відбираючи предгон з більш низькою температурою кипіння. Вихід продукту складає 11,2 г (54,2 % від теоретичного).
Отриманий продукт перевіряють по температурі кипіння: Ттопл.
= 35°С, Ткип.
= 164,5°С (літ. Ттопл.
= 35,5°С, Ткип.
= 163,8°С) [10].
2.1.8 Етоксіоцтова кислота
Синтез етоксіоцтової кислоти здійснюють за наступною схемою [16]:

У дву горлу колбу об’ємом 250 мл, що обладнана зворотним холодильником довжиною 70-80 см із хлоркальциєвою трубкою, наливають 180 мл абсолютного етилового спирту. Через бічне горло поступово додають 10 г металічного натрію протягом 1 години. Потім у бічне горло колби вміщують крапельну воронку з 20,3 г (0,215 моль) хлороцтовї кислоти в 26 мл абсолютного етилового спирту і повільно прикапують спиртовий розчин хлороцтової кислоти у розчин етилату натрію протягом 15-20 хвилин. Суміш нагрівають на водяній бані протягом 10 хвилин і відганяють 150 мл етанолу. Після цього доливають 250 мл дистильованої води і відганяють на водяній бані приблизно 100 мл рідини, у якій міститься приблизно 20 мл етанолу (перевіряють за показником заломлення: nD
=1,3645, а для етанолу nD
=1,3614). Відгонку продовжують доти, поки за показником заломлення не буде відганятися чиста вода. Водяний розчин, що залишився в колбі, охолоджують і додають до нього 45 мл концентрованої соляної кислоти (HCl, ρ= 1,19 г/мл). Хлорид натрію, що випав, відфільтровують на воронці Бюхнера і промивають два рази порціями по 10 мл диетилового ефіру. Ефірний шар відокремлюють від водяного. Водяний шар потім екстрагують ще чотири рази ефіром порціями по 30 мл. Ефірні витяжки поєднують і насичують їх 15 г сірчанокислого натрію Na2
SO4
. Ефір відганяють на водяній бані. Отриману кислоту переганяють при зниженому тиску. Вихід продукту складає 9,1 г (40,7 % від теор.).
Отриманий продукт перевіряють по температурі кипіння: Ткип.
= 207,5°С, nD
= 1,4191 (літ. Ткип.
= 206-207°С з частковим розкладанням, nD
= 1,4194) [10].
2.1.9 Феноксіоцтова кислота
Синтез феноксіоцтової кислоти здійснюють за наступною схемою [20]:
C6
H5
ONa + ClCH2
COONa C6
H5
OCH2
COONa + NaCl C6
H5
OCH2
COONa + NaCl
C6
H5
OCH2
COONa + HCl C6
H5
OCH2
COOH + NaCl
У колбі, що обладнана зворотним холодильником, змішують 3,0 г фенолу (0,032 моль), 30 мл 25%-го розчину гідроксиду натрію (0,4 моль) та 7,2 г (0,076 моль) хлороцтової кислоти. Суміш нагрівають на киплячій водяній бані протягом 1 години, а потім охолоджують, підкислюють 18 мл 10%-ою соляної кислоти до рН=3─5 (по конго червоному) та екстрагують двічі ефіром порціями по 16 мл. Ефірні витяжки обережно змішують у стакані з 40 мл розчину карбонату натрію. Після припинення виділення вуглекислого газу суміш встряхують у ділільній воронці. Ефірний шар відділяютьють, а водний – повільно та обережно при розмішуванні у стакані підкислюють концентрованою соляною кислотою до рН=3─5 по конго червоному. Феноксіоцтову кислоту, що випала, відфільтровують, промивають на фільтрі 6 мл води та висушують на повітрі.
Вихід складає 1,2 г (25% від теоретично можливого).
Феноксіоцтову кислоту перекристалізовують з води (на 1 г речовини беруть 75 мл води), отримують безбарвні голки; Ттопл.
= 90-910
С (літ. Ттопл.
= 91°С) [10].
2.1.10 Фенілоцтова кислота
Синтез фенілоцтової кислоти здійснюють за наступною схемою [21]:
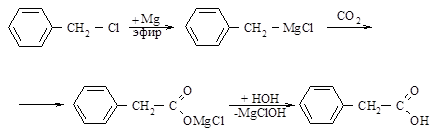
У круглодонну колбу на 250 мл вміщують 2,4 г (0,1 моль) магнію, додають кілька кристалів йоду та нагрівають до виникнення забарвлення (2 – 3 хвилини). Потім у колбу, що обладнана дворогим форштосом із вставленими в нього крапельною воронкою та зворотним холодильником з хлоркальцієвою трубкою, доливають 30 мл абсолютного етилового ефіру та додають 3 – 4 мл розчину 12,7 г (0,1 моль) хлористого бензилу у 50 мл абсолютного ефіру. Після того, як реакція почнеться, ефір помутніє та закипить (використовується баня з теплою водою), додають по краплям ефірний розчин хлористого бензилу з такою швидкістю, щоб ефір увесь час спокійно та рівномірно кипів. Після введення усього хлористого бензилу суміш нагрівають на водяній бані 1 – 1,5 години до повного розчинення магнію. Охолоджують колбу сумішшю льоду та солі (до 50
С), замінюють крапельну воронку газоприводною трубкою і пропускають протягом 3 – 4 годин не занадто сильний струм сухого СО2
з апарата Кипа, висушуючи його пропущенням через дві склянки Тищенка із концентрованою сірчаною кислотою. Потім замінюють газоприводну трубку крапельною воронкою і при сильному охолодженні (до 50
С) та розмішуванні додають по краплях розчин 10 г концентрованої соляної кислоти (ρ=1,19 г/мл) у 20 мл води. Після того, як реакційна суміш розділиться на два прозорих шари, вміст колби переносять у ділільну воронку, добре струшують та після відстоювання відокремлюють водяний шар. Водяний шар двічі екстрагують ефіром (по 20 мл). Об’єднані ефірні розчини фенілоцтової кислоти повторно струшують з 50 мл 20% розчину NaOH, лужний розчин відокремлюють, підкислюють 50 мл 10% розчину HCl і охолоджують.
Отриману фенілоцтову кислоту відфільтровують, промивають холодною водою, добре віджимають та перекристалізовують з гарячої води.
Вихід складає 3,2 г (38,1% від теоретично можливого). Ттопл.
=750
С (літ. Ттопл.
= 74 - 75°С) [10].
2.1.11 Масляна кислота
Масляну кислоту очищують перегонкою при атмосферному тиску, збираючи фракції з Ткип
= 163-164°С, nD
= 1,3975 (літ. Ткип
= 163,5°С, nD
= 1,3979) [10].
2.1.12 Ізомасляна кислота
Ізомасляну кислоту очищують перегонкою при атмосферному тиску, збираючи фракції з Ткип
= 154-155°С, nD
= 1,3925 (літ. Ткип
= 154,5°С, nD
= 1,3930) [10].
2.1.13 Пропіонова кислота
Пропіонова кислота, що випускається у промисловості, містить домішки карбонільних сполук, що віддаляються кип’ятінням зі зворотним холодильником у присутності 5 вагових відсотків KMnO4
протягом 1 години, після чого кислоту переганяють. Залишки води видаляють з кислоти перегонкою при атмосферному тиску над Р2
О5
: Ткип
= 114-115°С, nD
= 1,3871 (літ. Ткип
= 141,1°С, nD
= 1,3874) [10].
2.2 Методика кінетичних вимірювань
Необхідні розчини реагентів готують за точною наважкою речовин. Точну концентрацію кислотного реагенту встановлюють за допомогою кислотно-основного потенціометричного титрування.
До однієї частини кінетичної колби вносять 2 мл розчину карбонової кислоти в епіхлоргідрині, а до другої – 1 мл розчину каталізатору в епіхлоргідрині. Колбу термостатують протягом 10 хвилин, а потім змішують розчини з обох частин кінетичної колби, точно відмічаючи при цьому час початку реакції. Через заданий проміжок часу реакцію припиняють шляхом доливання до реакційної суміші 15 мл охолодженої суміші ізопропілового спирту та води (1: 1) при швидкому змішуванні.
Вміст колби кількісно переносять водою до стакану для титрування та титрують лугом (NaOH), визначаючи поточну концентрацію кислоти.
Кількість кислоти, що не прореагувала, розраховують за рівнянням:
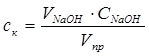 (2.1), (2.1),
де 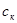 - концентрація карбонової кислоти, моль/л; - концентрація карбонової кислоти, моль/л;
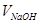 - об’єм розчину NaOH, який має концентрацію - об’єм розчину NaOH, який має концентрацію 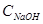 , що пішов на титрування, мл; , що пішов на титрування, мл;
 - - об’єм проби реакційної суміші, мл. - - об’єм проби реакційної суміші, мл.
2.3 Математична обробка експериментальних даних
Константи швидкості, які ми спостерігаємо, були розраховані за рівнянням нульового та першого порядків, виходячи з припущення, що реакція має нульовий порядок за кислотою, за формулами [17,18]:
для n=0 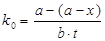 (2.2) (2.2)
для n=1  (2.3) (2.3)
де а – вихідна концентрація карбонової кислоти, моль/л;
(а-х) – поточна концентрація карбонової кислоти, моль/л;
t – час перебігу реакції, с;
b – вихідна концентрація епіхлоргідрина, моль/л;
Реакцію проводили в умовах псевдопорядку 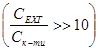 , що дозволяє знехтувати зміною концентрації епіхлоргідрину. , що дозволяє знехтувати зміною концентрації епіхлоргідрину.
Для статистичної оцінки констант швидкості використовували формулу [18]:
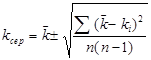 (2.4), (2.4),
де 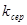 - середньоквадратичнаконстанта швидкості, с-1
; - середньоквадратичнаконстанта швидкості, с-1
;
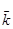 - середньоарифметична константа швидкості, с-1
; - середньоарифметична константа швидкості, с-1
;
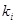 - і-те значення константи швидкості, с-1
; - і-те значення константи швидкості, с-1
;
 - кількість дослідів. - кількість дослідів.
Енергія активації Еа
була розрахована за формулою [17]:
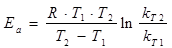 (2.5), (2.5),
де R - універсальна газова стала;
T1
, T2
- температура проведення реакції, К (Т2
>Т1
);
kТ1
, kT2
- костанти швидкості реакції при температурах Т1
та Т2
відновідно.
Передекспонентний множник А визначається з рівняння Ареніуса [17]:
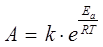 (2.6) (2.6)
Ентальпію активації DHТ
#
розраховували за формулою [17]:
DHТ
#
= Ea
-n×R×T (2.7),
деn - молекулярність реакції;
Ентропія активації DSТ
#
була розрахована за формулою [17]:
DSТ
#
= R×(ln A - ln T - 25,76) (2.8)
За правилом Вант-Гофа визначається температурний коефіцієнт γ [17]:
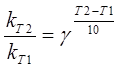 (2.9) (2.9)
Експериментальні дані були оброблені за рівняннями (2.2) – (2.9) з використанням сучасних комп’ютерних програм.
2.4 Техніка безпеки
Багато з органічних та неорганічних речовин, що використовуються в роботі, здійснюють шкідливий вплив на організм людини. Для безпечного використання роботи треба додержуватись правил техніки безпеки [19].
2.4.1 Робота з епіхлоргідрином [19]
Епіхлоргідрин – високотоксична речовина, яка має сильну подразнюючу дію. Проникаючи крізь дихальні шляхи та шкіру, викликає шкіряні захворювання. Всі роботи з епіхлоргідрином слід проводити в гумових рукавичках, у витяжній шафі.
При попаданні епіхлоргідрина на шкіру слід змити теплою водою з милом та протерти спиртом.
2.4.2 Робота з оцтовою кислотою [19]
Оцтова кислота подразнює шкіру, а також слизові оболонки дихальних шляхів. Пари викликають кашель та нежить, іноді тошноту та блювання. Роботу проводять у витяжній шафі.
2.4.3 Робота з амінами [19]
Токсична дія N,N-диметиланілінів подібнодо аніліну, але слабкіше. У легких випадках — синюха, невелика слабість, розбитість, головний біль, поганий апетит. Усі роботи з ними необхідно проводити у гумових рукавичках, нарукавниках, фартухах, чоботах, у костюмах з бавовняної тканини.
2.4.4 Робота з кислотами та лугами [19]
При попаданні кислот або лугів на шкіру необхідно промивання водою, пов'язки з 2-3% розчином соди у випадку попадання кислот або борної кислоти у випадку лугів.
Робота з даними речовинами проводиться у витяжній шафі, в гумових рукавицях, бо можливо пошкодження шкіри і виникнення дерматитів.
3. Результати та їх обговорення
Основною задачею даної роботи є встановлення впливу структури кислотного реагенту, концентрації та структури каталізатора, а також температури на швидкість ацидолізу епіхлоргідрину. Для вирішення цих питань було проведено цілеспрямоване варіювання структури карбонової кислоти, структури та концентрації каталізатора, а також варіювання температури в досліджуваній реакції (1).
Реакції ацидолізу епіхлоргідрину оцтовою кислотою та її похідними виконано в інтервалі температур 300
- 600
С при концентрації каталізатору від 0,00125 М до 0,005 М.
Дослідження проводилися з використанням розведених розчинів карбонових кислот в епіхлоргідрині (0,200 М) для виключення процесу само- асоціації. Усі експерименти виконані у середовищі сухого 1-хлор-2,3-епоксіпропану (W% Н2
О < 1%), що виключає можливість каталіза водою. За протіканням реакції слідкували за зменшенням кислотного реагенту методом кислотно-основного потенціометричного титрування.
Водночас було вирішено питання щодо порядку реакції за карбоновою кислотою та каталізатором в даних умовах.
3.1. Визначення порядку реакції за карбоновою кислотою.
Порядок реакції за кислотою було встановлено за допомогою метода сталості констант швидкості n-порядку при Т=333К.
Отримані результати наведені у табл. 3.1 – 3.6, де представлені константи швидкості, що розраховані як за першим, так і за нульовим порядками при Т=333К.
Спостерігаємі константи швидкості, які розраховані за рівнянням псевдопершого порядку, зростають за ходом процесу.
Константи швидкості псевдо нульового порядку є сталими у процесі протікання реакції.
Виходячи з цього, можна стверджувати, що реакція має нульовий порядок за карбоновою кислотою.
Таблиця 3.1 - Кінетика реакції (СН3
)3
ССООН (a)з ЕХГ (b=12,38 - 12,39 M) у присутності ДМА при Т=333К
| t, с |
(a-x), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
5400
7800
10200
12600
15000
17280
|
0,180
0,149
0,116
0,096
0,063
0,037
|
22,75
36,05
50,21
58,76
72,83
84,12
|
7,92
8,69
9,25
8,38
9,13
9,15
|
3,86
4,63
5,52
5,68
7,07
8,60
|
m=0,005 M; a=0,233 M; ρ=1,1707г/мл; b=12,39 M;
<k>=(8,75±0,18)·10-7
, с-1
|
3600
9000
11700
15300
17460
19500
|
0,199
0,152
0,119
0,088
0,063
0,050
|
14,59
34,76
48,93
62,23
72,96
78,54
|
7,62
7,26
7,65
7,86
7,57
7,86
|
3,54
3,83
4,64
5,14
6,05
6,37
|
m=0,00375 M; a=0,233 M; ρ=1,1699г/мл; b=12,38 M;
<k>=(7,64±0,08)·10-7
, с-1
|
3600
7200
10800
14400
18000
21600
|
0,208
0,181
0,147
0,120
0,083
0,059
|
10,73
22,32
36,91
48,50
64,38
74,68
|
5,60
5,83
6,43
6,28
6,73
6,50
|
2,55
2,83
3,44
3,72
4,63
5,14
|
m=0,0025 M; a=0,233 M; ρ=1,1697г/мл; b=12,38 M;
<k>=(6,24±0,15)·10-7
, с-1
|
4800
9000
13200
16800
20700
24600
|
0,199
0,172
0,146
0,124
0,106
0,081
|
14,59
26,18
37,34
46,78
54,51
65,41
|
5,71
5,47
5,31
5,23
4,95
5,00
|
2,65
2,72
2,86
3,03
3,07
3,47
|
m=0,00125 M; a=0,233 M; ρ=1,1703г/мл; b=12,39 M;
<k>=(5,28±0,10)·10-7
, с-1
|
Таблиця 3.2 - Кінетика реакції СН3
СН2
СООН (a)з ЕХГ (b=12,45 – 12,49 M) у присутності ДМА при Т=333К
| t, с |
(a-x), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
4260
8400
12600
16800
21000
25200
|
0,186
0,163
0,137
0,103
0,070
0,037
|
15,07
25,57
37,44
52,97
68,17
83,33
|
6,20
5,34
5,21
5,53
5,69
5,80
|
2,82
2,98
3,07
3,60
4,35
5,65
|
m=0,005 M; a=0,219 M; ρ=1,1715г/мл; b=12,48 M;
<k>=(5,63±0,12)·10-7
, с-1
|
4800
9600
14400
19260
24060
28800
|
0,195
0,171
0,142
0,115
0,080
0,049
|
10,96
21,92
35,16
47,49
63,47
77,63
|
4,01
4,01
4,29
4,33
4,64
4,74
|
1,94
2,07
2,42
2,67
3,36
4,18
|
m=0,00375 M; a=0,219 M; ρ=1,1685г/мл; b=12,45 M;
<k>=(4,34±0,11)·10-7
, с-1
|
5400
9060
16200
25200
29700
34200
|
0,195
0,183
0,156
0,114
0,090
0,073
|
10,96
16,44
28,77
47,95
58,90
66,67
|
3,55
3,18
3,11
3,33
3,47
3,41
|
1,59
1,68
1,72
2,07
2,40
2,57
|
m=0,0025 M; a=0,219 M; ρ=1,1723г/мл; b=12,49 M;
<k>=(3,34±0,06)·10-7
, с-1
|
6000
12000
18540
24660
32880
40560
45000
|
0,198
0,185
0,165
0,148
0,121
0,098
0,091
|
9,59
15,53
24,66
32,42
44,75
55,25
58,45
|
2,80
2,27
2,33
2,31
2,39
2,39
2,28
|
1,13
1,23
1,28
1,35
1,45
1,57
1,59
|
m=0,00125 M; a=0,219 M; ρ=1,1695г/мл; b=12,46 M;
<k>=(2,40±0,06)·10-7
, с-1
|
Таблиця 3.3 - Кінетика реакції СН3
СН(СН3
)СООН (a)з ЕХГ (b=12,46 - 12,52 M) у присутності ДМА при Т=333К
| t, с |
(a-x), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
3360
6300
9600
12600
15900
19320
|
0,154
0,130
0,104
0,079
0,053
0,025
|
14,92
28,18
42,54
56,35
70,72
86,19
|
6,48
6,53
6,47
6,53
6,49
6,51
|
3,88
4,24
4,66
5,31
6,23
8,27
|
m=0,005 M; a=0,181 M; ρ=1,1753г/мл; b=12,52 M;
<k>=(6,50±0,01)·10-7
, с-1
|
4440
7620
10920
14700
18600
23400
|
0,152
0,130
0,109
0,083
0,059
0,027
|
16,02
28,18
39,78
54,14
67,40
85,08
|
5,27
5,40
5,32
5,38
5,29
5,31
|
3,18
3,51
3,75
4,28
4,87
6,57
|
m=0,00375 M; a=0,181 M; ρ=1,1733г/мл; b=12,50 M;
<k>=(5,33±0,02)·10-7
, с-1
|
3660
8280
12480
18060
22860
28320
|
0,162
0,136
0,114
0,085
0,059
0,030
|
10,50
24,86
37,02
53,04
67,40
83,43
|
4,19
4,38
4,33
4,29
4,31
4,30
|
2,45
2,79
2,99
3,38
3,96
5,13
|
m=0,0025 M; a=0,181 M; ρ=1,1711г/мл; b=12,48 M;
<k>=(4,30±0,02)·10-7
, с-1
|
4800
8880
13080
18600
24000
28500
|
0,160
0,144
0,125
0,103
0,081
0,062
|
11,60
20,44
30,94
43,09
55,25
65,75
|
3,53
3,36
3,45
3,38
3,36
3,37
|
2,07
2,08
2,28
2,45
2,70
3,03
|
m=0,00125 M; a=0,181 M; ρ=1,1693г/мл; b=12,46 M;
<k>=(3,40±0,02)·10-7
, с-1
|
Таблиця 3.4 - Кінетика реакції PhOCH2
СООН (a)з ЕХГ (b=12,35 - 12,42 M) у присутності ДМА при Т=333К
| t, с |
(a-x), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
3600
7380
11820
13620
16800
18000
|
0,164
0,126
0,084
0,065
0,034
0,022
|
18,41
37,13
58,21
67,66
83,08
89,05
|
8,27
8,18
7,97
8,04
8,00
8,00
|
4,55
5,10
5,95
6,68
8,52
9,90
|
m=0,005 M; a=0,201 M; ρ=1,1799г/мл; b=12,42 M;
<k>=(8,08±0,04)·10-7
, с-1
|
3660
6600
11940
13920
16680
21960
|
0,169
0,143
0,099
0,085
0,069
0,022
|
15,92
28,86
50,75
57,71
65,67
89,05
|
7,04
7,08
6,88
6,71
6,37
6,56
|
3,82
4,16
4,78
4,98
5,17
8,12
|
m=0,00375 M; a=0,201 M; ρ=1,1793 г/мл; b=12,41 M;
<k>=(6,77±0,10)·10-7
, с-1
|
4200
8400
12600
16800
21000
24300
|
0,173
0,147
0,123
0,097
0,076
0,057
|
13,93
26,87
38,81
51,74
62,19
71,64
|
5,38
5,19
4,99
4,99
4,80
4,78
|
2,88
3,01
3,15
3,50
3,74
4,19
|
m=0,0025 M; a=0,201 M; ρ=1,1766 г/мл; b=12,38 M;
<k>=(5,02±0,08)·10-7
, с-1
|
9600
14400
19200
24000
28800
|
0,161
0,143
0,128
0,112
0,098
|
19,90
28,86
36,32
44,28
52,74
|
3,37
3,26
3,07
3,00
2,98
|
1,87
2,00
2,92
2,97
3,09
|
m=0,00125 M; a=0,201 M; ρ=1,1730 г/мл; b=12,35 M;
<k>=(3,27±0,14)·10-7
, с-1
|
Таблиця 3.5 - Кінетика реакції EtOCH2
СООН (a)з ЕХГ (b=12,50 - 12,53 M) у присутності ДМА при Т=333К
| t, с |
(a-x), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
3600
7200
10800
14400
18000
21600
|
0,159
0,134
0,106
0,078
0,051
0,023
|
14,97
28,34
43,32
58,29
72,73
87,70
|
6,20
5,87
5,98
6,04
6,03
6,06
|
3,60
3,69
4,20
4,85
5,76
7,74
|
m=0,005 M; a=0,187 M; ρ=1,1793г/мл; b=12,53 M;
<k>=(6,03±0,04)·10-7
, с-1
|
4800
9600
14400
19200
24000
27000
|
0,159
0,128
0,103
0,072
0,040
0,025
|
14,97
31,55
44,92
61,50
78,61
86,63
|
4,66
4,91
4,66
4,78
4,89
4,79
|
2,70
3,15
3,31
3,97
5,13
5,95
|
m=0,00375 M; a=0,187 M; ρ=1,1780 г/мл; b=12,52 M;
<k>=(4,78±0,04)·10-7
, с-1
|
3600
9000
12600
18000
25200
28500
|
0,170
0,147
0,126
0,099
0,062
0,050
|
9,09
21,39
32,62
47,06
66,84
73,26
|
3,53
3,88
3,82
3,85
3,94
3,75
|
2,12
2,14
2,51
2,82
3,50
3,70
|
m=0,0025 M; a=0,187 M; ρ=1,1770 г/мл; b=12,51 M;
<k>=(3,78±0,05)·10-7
, с-1
|
7200
10800
16200
19800
25320
32400
|
0,169
0,155
0,139
0,126
0,107
0,086
|
9,63
17,11
25,67
32,62
42,78
54,01
|
2,00
2,37
2,52
2,37
2,46
2,49
|
1,12
1,39
1,47
1,60
1,76
1,92
|
m=0,00125 M; a=0,187 M; ρ=1,1758 г/мл; b=12,50 M;
<k>=(2,37±0,07)·10-7
, с-1
|
Таблиця 3.6 - Кінетика реакції карбонових кислот (а=0,170 – 0,205 М)з ЕХГ (b=12,46 - 12,52 M) у присутності Et4
NBr (m=0,005 М) при Т=333К
| (СН3
)3
ССООН |
| t, с |
(a-x), М |
у, % |
kсп
·106
, с-1
|
kсп
·105
, л/моль· с |
300
600
900
1200
1500
1800
|
0,152
0,129
0,111
0,090
0,073
0,056
|
10,59
24,12
34,71
47,06
57,06
67,06
|
4,79
5,46
5,24
5,32
5,17
5,06
|
2,98
3,67
3,78
4,23
4,50
4,93
|
a=0,170 M; ρ=1,1765г/мл; b=12,52 M;
<k>=(5,17±0,08)·10-6
, с-1
|
| СН3
СН(СН3
)СООН |
780
1560
2340
3120
3900
4680
|
0,172
0,146
0,123
0,098
0,074
0,055
|
11,79
25,13
36,92
49,74
62,05
71,79
|
2,37
2,52
2,47
2,50
2,49
2,40
|
1,29
1,49
1,58
1,77
1,99
2,17
|
a=0,195 M; ρ=1,1709г/мл; b=12,46 M;
<k>=(2,46±0,02)·10-6
, с-1
|
| СН3
СН2
СН2
СООН |
1205
2400
3600
4800
6000
7200
|
0,174
0,145
0,116
0,089
0,070
0,041
|
15,12
29,27
43,41
56,59
65,85
80,00
|
2,06
2,00
1,98
1,94
1,80
1,83
|
1,09
1,16
1,27
1,39
1,44
1,79
|
a=0,205 M; ρ=1,1730г/мл; b=12,47 M;
<k>=(1,94±0,03)·10-6
, с-1
|
| EtOCH2
СООН |
1500
1800
3000
4200
5400
6600
7800
|
0,163
0,151
0,129
0,102
0,069
0,040
0,016
|
18,09
24,12
35,18
48,74
65,33
79,90
91,96
|
1,92
2,14
1,87
1,85
1,93
1,93
1,88
|
1,07
1,16
1,23
1,28
1,57
1,95
2,59
|
a=0,199 M; ρ=1,1761г/мл; b=12,48 M;
<k>=(1,93±0,03)·10-6
, с-1
|
На основі отриманих експериментальних даних були побудовані графіки залежності концентрації кислоти від часу проведення реакції (рис. 3.1 – 3.6), які носять прямолінійний характер (r = 0,997 – 0,999), що також підтверджує нульовий порядок реакції за карбоновою кислотою.
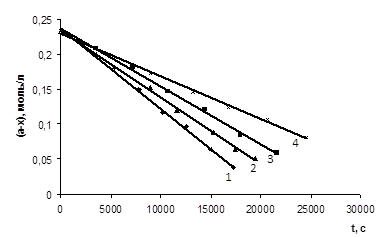
Рис. 3.1 Залежність концентрації (СН3
)3
ССООН від часу t реакції (СН3
)3
ССООН (а=0,233 М) з епіхлоргідрином (12,38 – 12,39 М) при різних концентраціях катализатора N, N-диметиланіліна (m = 0,00125 – 0,005 М) при Т=333К 1 – m=0,005 M; 2 – m=0,00375 M; 3 – m=0,0025 M; 4 – m=0,00125 M
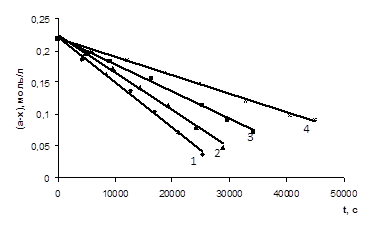
Рис. 3.2 Залежність концентрації СН3
СН2
СООН від часу t реакції СН3
СН2
СООН (а=0,233 М) з епіхлоргідрином (12,45 – 12,49 М) при різних концентраціях катализатора N, N-диметиланіліна (m = 0,00125 – 0,005 М) при Т=333К 1 – m=0,005 M; 2 – m=0,00375 M; 3 – m=0,0025 M; 4 – m=0,00125 M
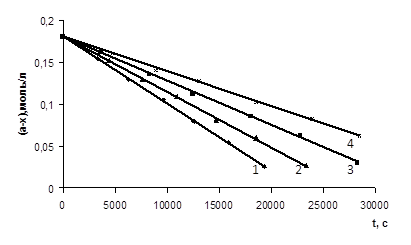
Рис. 3.3 Залежність концентрації СН3
СН(СН3
)СООН від часу t реакції СН3
СН(СН3
)СООН (а=0,181 М) з епіхлоргідрином (12,46 – 12,52 М) при різних концентраціях катализатора N, N-диметиланіліна (m = 0,00125 – 0,005 М) при Т=333 К1 – m=0,005 M; 2 – m=0,00375 M; 3 – m=0,0025 M; 4 – m=0,00125 M
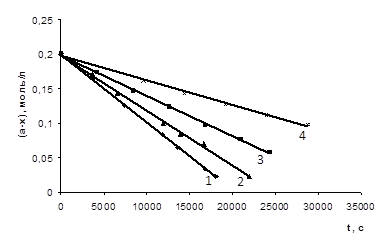
Рис. 3.4 Залежність концентрації PhOСН2
СООН від часу t реакції PhOСН2
СООН (а=0,201 М) з епіхлоргідрином (12,35 – 12,42 М) при різних концентраціях катализатора N, N-диметиланіліна (m = 0,00125 – 0,005 М) при Т=333К 1 – m=0,005 M; 2 – m=0,00375 M; 3 – m=0,0025 M; 4 – m=0,00125 M
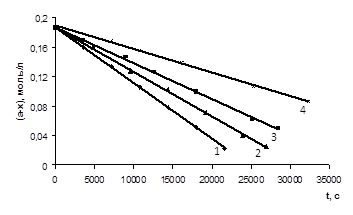
Рис. 3.5 Залежність концентрації EtOСН2
СООН від часу t реакції EtOСН2
СООН (а=0,201 М) з епіхлоргідрином (12,50 – 12,53 М) при різних концентраціях катализатора N, N-диметиланіліна (m = 0,00125 – 0,005 М) при Т=333К 1 – m=0,005 M; 2 – m=0,00375 M; 3 – m=0,0025 M; 4 – m=0,00125 M
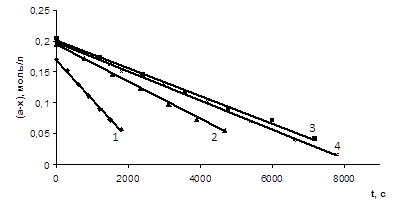
Рис. 3.6 Залежність концентрації карбонової кислоти від часу t реакції (СН3
)3
ССООН (а=0,170 М), СН3
СН(СН3
)СООН (а=0,195 М), СН3
СН2
СН2
СООН (а=0,205 М), EtOCH2
СООН (а=0,199 М) з епіхлоргідрином (12,46 – 12,52 М) при концентрації катализатору Et4
NBr (m=0,005 М) при Т=333К 1 - (СН3
)3
ССООН; 2 - СН3
СН(СН3
)СООН; 3 - СН3
СН2
СН2
СООН; 4 - EtOCH2
СООН
3.2 Визначення порядку реакції за каталізатором
Порядок реакції за каталізатором досліджувався у реакції триметилоцтової, ізомасляної, пропіонової, феноксі- та етоксіоцтової кислот з епіхлоргідрином у присутності каталізатору N, N-диметиланіліна при Т=333К, а також у реакції оцтової кислоти з епіхлоргідрином у присутності тетраетиламонію броміду, γ-піколіну, 4-Br-N,N-диметиланіліну та 3-NO2
-N,N-диметиланіліну при Т=333К. Концентрація каталізатора варіювалася в інтервалі 0,00125 – 0,005 М.Отримані результати наведені у табл. 3.7, 3.8, де представлені константи швидкості, які отримані при варіюванні вище зазначених похідних оцтової кислоти та каталізатору при різних концентраціях останнього.
Таблиця 3.7 - Залежність спостерігаємої константи швидкості карбонових кислот від концентрації (m) каталізатора N, N-диметиланіліна
| Кислота |
m, моль/л |
kсп
·107
, с-1
|
k0
·107
, с-1
|
kкат
·104
, л/моль·с |
r |
| (СН3
)3
ССООН |
0,00500
0,00375
0,00250
0,00125
|
8,75±0,18
7,64±0,08
6,24±0,15
5,28±0,10
|
4,03±0,16 |
0,945±0,045 |
0,998 |
| СН3
СН2
СООН |
0,00500
0,00375
0,00250
0,00125
|
5,63±0,12
4,34±0,11
3,34±0,06
2,40±0,06
|
1,26±0,16 |
0,855±0,046 |
0,997 |
| СН3
СН(СН3
)СООН |
0,00500
0,00375
0,00250
0,00125
|
6,50±0,01
5,33±0,02
4,30±0,02
3,40±0,02
|
2,30±0,12 |
0,826±0,034 |
0,998 |
| Ph-O-CH2
COOH |
0,00500
0,00375
0,00250
0,00125
|
8,08±0,04
6,77±0,10
5,02±0,08
3,27±0,14
|
1,74±0,21 |
1,29±0,06 |
0,998 |
| Et-O-CH2
COOH |
0,00500
0,00375
0,00250
0,00125
|
6,03±0,04
4,78±0,04
3,78±0,05
2,37±0,07
|
1,25±0,15 |
0,96±0,04 |
0,998 |
Таблиця 3.8 - Залежність спостерігаємої константи швидкості оцтової кислоти від концентрації (m) каталізаторів
| Каталізатор |
m, моль/л |
kсп
·107
, с-1
|
k0
·107
, с-1
|
kкат
·104
, л/моль·с |
r |
| Et4
NBr |
0,00500
0,00375
0,00250
0,00125
|
11,00±0,20
8,25±0,09
5,50±0,15
2,75±0,07
|
0,005±0,002 |
2,20±0,03 |
0,999 |
| 4-Me-Py |
0,00500
0,00375
0,00250
0,00125
|
5,50±0,11
4,13±0,11
2,75±0,04
1,38±0,02
|
0,005±0,002 |
1,10±0,05 |
0,999 |
| ДМА |
0,00500
0,00375
0,00250
0,00125
|
4,20±0,11
3,54±0,08
2,89±0,08
2,00±0,03
|
1,35±0,11 |
0,580±0,032 |
0,997 |
| 4-Br-ДМА |
0,00500
0,00375
0,00250
0,00125
|
1,58±0,01
1,19±0,02
0,79±0,01
0,40±0,01
|
0,005±0,002 |
0,315±0,001 |
0,999 |
| 3-NO2
-ДМА |
0,00500
0,00375
0,00250
0,00125
|
0,66±0,02
0,50±0,10
0,33±0,03
0,17±0,01
|
0,005±0,002 |
0,131±0,001 |
0,999 |

Рис. 3.7 Залежність kсп
від концентрації каталізатора (m) N, N-диметиланіліна для реакції карбонових кислот (0,181 – 0,233 М) з епіхлоргідрином (12,35 – 12,53 М) при Т=333 К 1 - (СН3
)3
ССООН; 2 - СН3
СН2
СООН; 3 – СН3
СН(СН3
)СООН; 4 – Ph-O-CH2
COOH; 5 – Et-O-CH2
COOH

Рис. 3.8 Залежність kсп
від концентрації каталізатора (m) реакції оцтової кислоти (0,192 – 0,210 М)з епіхлоргідрином (12.55 – 12.62 М) при Т=333 К
1 - Et4
NBr; 2 - 4-Me-Py; 3 - 4-Br-ДМА; 4 - 3-NO2
-ДМА
Прямолінійність отриманих ліній для різних карбонових кислот та каталізаторів вказує на перший порядок реакції за каталізатором. При цьому встановлено, що швидкість некаталітичного потоку реакції значно менше швидкості каталітичного потоку. Таким чином, встановлено, що тетраетиламоній бромід, γ-піколін, N,N-диметиланілін, 4-Br-N,N-диметиланілін та 3-NO2
-N,N-диметиланілін є ефективними каталізаторами ацидолізу епіхлоргідрину. Залежність Бренстеда (рис. 3.9) має прямолінійний характер, а коефіцієнт β=(0,279±0,015) вказує на низьку чутливість реакції до основності каталізатору, яка характерна для загальноосновного каталізу [23].
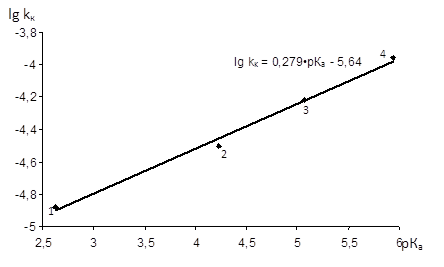
Рис. 3.9 Залежність lg kсп
від рКа
каталізатора (m=0,005 М) реакції оцтової кислоти (0,197 - 0,210 М) з ЕХГ (12,55 - 12,62 М) при Т=333К 1 - 3-NO2
-ДМА; 2 - 4-Br-ДМА; 3 - ДМА; 4 - 4-Me-Py
3.3 Вплив структури кислотного реагенту на швидкість ацидолізу епіхлоргідрину
Для оцінки впливу структури кислотного реагенту було проварійовано природу замісника в аліфатичних монокарбонових кислотах (табл. 3.9).
Таблиця 3.9 - Залежність спостерігаємої константи швидкості ацидолызу ЕХГ в присутності N,N-диметиланіліну від константи замісника σ*
в карбоновій кислоті
| Кислота |
σ*
|
kсп
·107
, с-1
|
pKa
|
(СН3
)3
ССООН
СН3
СН(СН3
)СООН
СН3
СН2
СООН
СН3
СООН
Ph-CH2
COOH
Et-O-CH2
COOH
Ph-O-CH2
COOH
|
-0,30
-0,19
-0,10
0
0,215
0,65
0,85
|
8,75±0,18
6,50±0,01
5,63±0,12
4,20±0,11
5,71±0,08
6,03±0,04
8,08±0,04
|
5,03
4,85
4,87
4,75
4,31
3,55
3,17
|
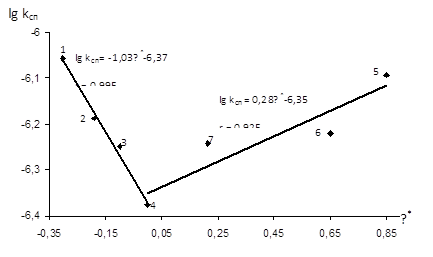
Рис.3.10 Залежність lgkсп
від σ*
замісника в оцтовій кислоті (0,181 – 0,233 М) при ацидолізі ЕХГ (12,35 – 12,53 М) у присутності C6
H5
N(CH3
)2
(m=0,005 М) при Т=333К 1- (СН3
)3
ССООН; 2- СН3
СН(СН3
)СООН; 3- СН3
СН2
СООН; 4-CH3
COOH; 5- Ph-O-CH2
COOH; 6 – Et-O-CH2
COOH; 7 - Ph-CH2
COOH
Прямолінійна залежність у координатах lg kсп
від σ*
у випадку похідних оцтової кислоти з електронодонорними замісниками показує, що із збільшенням кислотних властивостей –ОН реагенту швидкість реакції зменшується, а отриманий коефіцієнт ρ1
*
=-1,03 вказує на збільшення швидкості реакції ацидолізу епіхлоргідрину із підвищенням донорних властивостей замісника в оцтовій кислоті, тобто із збільшенням ефективного негативного заряду на атомі кисню карбоксильної групи.
У випадку електроноакцепторних замісників в оцтовій кислоті також спостерігається прямолінійна залежність у координатах lg kсп
─ σ*
, а коефіцієнт ρ2
*
=0,28, який вказує на збільшення швидкості реакції із збільшенням кислотних властивостей карбонової кислоти.
З наведених значень ρ1
*
та ρ2
*
видно, що чутливість реакційної серії до зміни електронодонорного замісника є значно вищою, ніж чутливість до зміни замісника електроноакцепторного характеру.
Для оцінки впливу сили кислотного реагенту на швидкість ацидолізу епіхлоргідрину була також побудована залежність у координатах lg kсп
─ рКа
кислоти (рКа
=3,17 ─ 5,03):
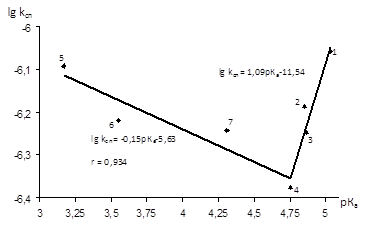
Рис.3.11 Залежність lgkсп
від рКа
кислоти при ацидолізі ЕХГ у присутності C6
H5
N(CH3
)2
(m=0.005 М) при Т=333К 1- (СН3
)3
ССООН; 2- СН3
СН(СН3
)СООН; 3- СН3
СН2
СООН; 4-CH3
COOH; 5- Ph-O-CH2
COOH; 6 – Et-O-CH2
COOH; 7 - Ph-CH2
COOH
Як і у випадку залежності Тафта, залежність Бренстеда носить V-подібний характер, але з гіршим коефіцієнтом кореляції. Коефіцієнт β=1,09±0,24 у випадку кислот з електронодонорними замісниками вказує на чутливість реакції до зміни сили кислоти з рКа
=4,75 ─ 5,03.
У випадку ж кислот із замісниками електроноакцепторної природи спостерігається дуже низька чутливість реакції ( β=-0,15±0,04) до сили кислот з рКа
=4,75 ─ 3,17.
Таким чином, такий вид ламаної прямої можна пояснити зміною швидкістьвизначаючої стадії при переході від електронодонорної до електроноакцепторної природи замісника в молекулі оцтової кислоти в рамках єдиного механізму реакції.
3.4 Вплив температури на швидкість ацидолізу епіхлоргідрину
Для оцінки впливу температури на швидкість ацидолізу ЕХГ булапроварійована температура досліджуваної реакції в інтервалі 30 – 600
С.
Отримані експериментальні дані наведені у табл. 3.10 – 3.18.
Таблиця 3.10 - Кінетика реакції СН3
СООН (а=0,198 М)з ЕХГ (b=12,55 М) у присутності каталізатора (C2
H5
)4
NBr (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
27000
57600
86600
118800
152160
|
0,173
0,145
0,120
0,090
0,062
|
12,63
26,77
39,39
54,55
68,69
|
0,746
0,733
0,720
0,728
0,712
|
0,398
0,431
0,461
0,529
0,608
|
| Т=303 К; <k>=(0,728±0,006)·10-7
, с-1
|
14400
21600
36000
50400
59400
|
0,163
0,147
0,114
0,080
0,060
|
17,68
25,76
42,42
59,60
69,70
|
1,96
1,89
1,86
1,86
1,85
|
1,08
1,10
1,22
1,43
1,60
|
| Т=313 К; <k>=(1,88±0,02)·10-7
, с-1
|
3600
7200
10920
15660
22080
|
0,175
0,151
0,126
0,098
0,058
|
11,62
23,74
36,36
50,51
70,71
|
5,18
5,17
5,29
5,07
5,07
|
2,73
3,00
3,30
3,58
4,43
|
| Т=323 К; <k>=(5,16±0,04)·10-7
, с-1
|
3000
4500
6000
7500
9000
|
0,151
0,128
0,106
0,083
0,065
|
23,74
35,35
46,46
58,08
67,17
|
12,40
12,40
12,10
12,10
11,70
|
7,20
7,72
8,30
9,24
9,86
|
| Т=333 К; <k>=(12,10±0,13)·10-7
, с-1
|
Таблиця 3.11 - Кінетика реакції СН3
СООН (а=0,197 М)з ЕХГ (b=12,61 М) у присутності каталізатора 4-Me-C5
H4
N (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
43380
72000
144720
180600
225000
244800
|
0,176
0,157
0,121
0,104
0,084
0,071
|
10,66
20,30
30,58
47,21
57,36
63,96
|
0,393
0,440
0,417
0,407
0,400
0,409
|
0,206
0,250
0,267
0,281
0,300
0,331
|
| Т=303 К; <k>=(0,411±0,007)·10-7
, с-1
|
16800
32400
48600
64800
81060
99780
|
0,173
0,154
0,133
0,112
0,092
0,070
|
12,18
21,83
32,49
43,15
53,30
64,47
|
1,14
1,06
1,04
1,04
1,03
1,01
|
0,603
0,613
0,641
0,691
0,745
0,822
|
| Т=313 К; <k>=(1,05±0,06)·10-7
, с-1
|
6600
13260
19800
26520
31200
37800
|
0,176
0,151
0,129
0,109
0,094
0,074
|
10,66
23,35
34,52
44,67
52,28
62,44
|
2,50
2,74
2,74
2,64
2,61
2,58
|
1,35
1,59
1,70
1,77
1,88
2,05
|
| Т=323 К; <k>=(2,63±0,04)·10-7
, с-1
|
3000
5700
8520
11100
13800
16200
|
0,169
0,147
0,125
0,109
0,086
0,071
|
14,21
25,38
36,55
44,67
56,35
63,96
|
7,41
7,02
6,73
6,34
6,43
6,25
|
4,09
4,12
4,27
4,28
4,81
5,05
|
| Т=333 К; <k>=(6,70±0,18)·10-7
, с-1
|
Таблиця 3.12 - Кінетика реакції СН3
СООН (а=0,200 М)з ЕХГ (b=12,57 М) у присутності каталізатора C6
H5
N(CH3
)2
(m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
38340
95220
172800
207240
252000
309600
|
0,183
0,161
0,127
0,112
0,089
0,065
|
8,50
19,50
36,50
44,00
55,50
67,50
|
0,360
0,323
0,343
0,339
0,351
0,346
|
1,88
1,79
2,07
2,24
2,56
2,87
|
| Т=303 К; <k>=(0,342±0,005)·10-7
, с-1
|
19860
39720
60720
79200
102600
118800
|
0,180
0,163
0,140
0,123
0,094
0,074
|
10,20
18,30
30,10
38,60
52,90
62,80
|
0,815
0,734
0,789
0,775
0,821
0,841
|
4,29
4,05
4,69
4,89
5,84
6,61
|
| Т=313 К; <k>=(0,796±0,016)·10-7
, с-1
|
7800
16320
23940
33060
43200
48600
|
0,182
0,161
0,145
0,120
0,093
0,077
|
8,86
19,31
27,50
39,90
53,60
61,50
|
1,81
1,88
1,83
1,92
1,97
2,01
|
9,46
10,5
10,7
12,3
14,2
15,6
|
| Т=323 К; <k>=(1,91±0,03)·10-7
, с-1
|
4260
8400
16800
21000
25620
|
0,176
0,160
0,110
0,091
0,061
|
11,50
19,20
42,70
52,00
65,90
|
4,50
3,82
4,25
4,14
4,30
|
20,2
22,8
26,4
27,8
33,4
|
| Т=333 К; <k>=(4,20±0,11)·10-7
, с-1
|
Таблиця 3.13 - Кінетика реакції СН3
СООН (а=0,210 М) з ЕХГ (b=12,59 М) у присутності каталізатора 4-Br-C6
H4
N(CH3
)2
(m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
46800
93600
16200
259200
334800
460800
|
0,198
0,186
0,169
0,145
0,119
0,083
|
5,71
11,43
19,52
30,95
43,33
60,48
|
0,199
0,205
0,201
0,199
0,215
0,219
|
0,100
0,103
0,106
0,113
0,135
0,160
|
| Т=303 К; <k>=(0,206±0,004)·10-7
, с-1
|
72000
90000
144600
195900
225000
|
0,167
0,153
0,108
0,081
0,059
|
20,48
27,14
48,57
61,43
71,90
|
0,474
0,500
0,561
0,025
0,534
|
0,253
0,279
0,365
0,386
0,448
|
| Т=313 К; <k>=(0,521±0,012)·10-7
, с-1
|
14460
28800
45000
57600
75600
86400
100800
|
0,187
0,170
0,146
0,120
0,110
0,081
0,059
|
10,95
19,05
30,48
42,86
47,62
61,43
71,90
|
1,29
1,11
1,13
1,24
1,16
1,19
1,19
|
0,583
0,637
0,642
0,680
0,772
0,876
1,00
|
| Т=323 К; <k>=(1,19±0,02)·10-7
, с-1
|
7200
15240
20400
34260
37800
49020
|
0,185
0,170
0,150
0,105
0,097
0,058
|
11,90
19,05
28,57
50,00
53,81
72,38
|
2,80
2,07
2,33
2,44
2,38
2,47
|
1,10
1,31
1,40
1,61
1,62
2,08
|
| Т=333 К; <k>=(2,42±0,10)·10-7
, с-1
|
Таблиця 3.14 - Кінетика реакції СН3
СООН (а=0,210 М) з ЕХГ (b=12,54 М) у присутності каталізатора 3-NO2
-C6
H4
N(CH3
)2
(m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·107
, л/моль· с |
111000
226980
423000
555000
819000
1020000
|
0,194
0,181
0,150
0,132
0,098
0,076
|
7,62
13,81
28,57
37,14
53,33
63,81
|
0,115
0,102
0,113
0,112
0,109
0,105
|
0,522
0,569
0,634
0,667
0,742
0,795
|
| Т=303 К; <k>=(0,109±0,002)·10-7
, с-1
|
97200
212400
280860
367200
453600
540000
|
0,195
0,158
0,133
0,117
0,095
0,069
|
7,14
24,76
36,67
44,29
54,76
67,14
|
0,120
0,194
0,218
0,201
0,202
0,209
|
0,608
1,07
1,27
1,30
1,39
1,64
|
| Т=313 К; <k>=(0,191±0,014)·10-7
, с-1
|
72000
108000
144000
181200
234000
|
0,183
0,159
0,147
0,122
0,096
|
12,86
24,29
30,00
41,90
54,29
|
0,301
0,376
0,348
0,387
0,387
|
1,52
1,98
2,05
2,39
2,67
|
| Т=323 К; <k>=(0,360±0,043)·10-7
, с-1
|
21600
57600
93600
115200
144360
|
0,193
0,162
0,131
0,105
0,070
|
8,10
22,86
37,62
50,00
66,67
|
0,638
0,669
0,676
0,728
0,773
|
0,31
0,36
0,40
0,48
0,61
|
| Т=333 К; <k>=(0,697±0,020)·10-7
, с-1
|
Таблиця 3.15 - Кінетика реакції (СН3
)3
ССООН (а=0,170 М)з ЕХГ (b=12,52 М) у присутності каталізатора (C2
H5
)4
NBr (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
6240
13620
19500
24000
30000
36000
|
0,151
0,112
0,090
0,075
0,052
0,036
|
11,18
34,12
47,06
55,88
69,41
78,82
|
2,43
3,40
3,28
3,16
3,14
2,97
|
1,52
2,45
3,61
2,72
3,15
3,44
|
| Т=303 К; <k>=(3,07±0,12)·10-7
, с-1
|
2400
4800
7200
9600
12000
14400
|
0,149
0,116
0,094
0,071
0,047
0,026
|
12,35
31,76
44,71
58,24
72,35
84,71
|
6,99
8,99
8,43
8,24
8,19
7,99
|
4,39
6,36
6,57
7,27
8,56
10,42
|
| Т=313 К; <k>=(8,14±0,27)·10-7
, с-1
|
900
1800
2700
3600
4500
5400
|
0,148
0,116
0,091
0,067
0,043
0,024
|
12,94
31,76
46,47
60,59
74,71
85,88
|
19,53
23,97
23,38
22,86
22,55
21,60
|
12,30
16,96
18,49
20,66
24,41
28,97
|
| Т=323 К; <k>=(22,31±0,55)·10-7
, с-1
|
300
600
900
1200
1500
1800
|
0,152
0,129
0,111
0,090
0,073
0,056
|
10,59
24,12
34,71
47,06
57,06
67,06
|
47,94
54,59
52,38
53,26
51,67
50,60
|
29,81
36,75
37,84
42,34
45,03
49,29
|
| Т=333 К; <k>=(51,74±0,80)·10-7
, с-1
|
Таблиця 3.16 - Кінетика реакції СН3
СН(СН3
)СООН (а=0,195 М)з ЕХГ (b=12,46 М) у присутності каталізатора (C2
H5
)4
NBr (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
23400
35100
47566
61260
72900
|
0,172
0,140
0,117
0,085
0,071
|
11,79
28,21
40,00
56,41
63,59
|
1,30
1,26
1,32
1,44
1,37
|
4,31
7,58
8,62
10,88
11,12
|
| Т=303 К; <k>=(1,34±0,02)·10-7
, с-1
|
4800
9600
14400
19200
24000
28800
|
0,184
0,153
0,126
0,103
0,078
0,061
|
5,64
21,54
35,38
47,18
60,00
68,72
|
3,77
3,51
3,85
3,85
3,91
3,73
|
0,971
2,03
2,43
2,67
3,06
3,24
|
| Т=313 К; <k>=(3,77±0,05)·10-7
, с-1
|
1920
3840
5760
7680
9600
11520
|
0,171
0,143
0,120
0,093
0,072
0,052
|
12,31
26,67
38,46
52,31
63,08
73,33
|
10,03
10,87
10,45
10,66
10,28
9,96
|
5,49
6,48
6,77
7,74
8,33
9,21
|
| Т=323 К; <k>=(10,38±0,12)·10-7
, с-1
|
780
1560
2340
3120
3900
4680
|
0,172
0,146
0,123
0,098
0,074
0,055
|
11,79
25,13
36,92
49,74
62,05
71,79
|
23,67
25,21
24,70
24,96
24,90
24,01
|
12,92
14,89
15,81
17,70
19,94
21,71
|
| Т=333 К; <k>=(24,58±0,21)·10-7
, с-1
|
Таблиця 3.17 - Кінетика реакції СН3
СН2
СН2
СООН (а=0,205 М)з ЕХГ (b=12,47 М) у присутності каталізатора (C2
H5
)4
NBr (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
27000
34200
54000
70380
86400
104400
|
0,166
0,159
0,134
0,110
0,087
0,069
|
19,02
22,44
34,63
53,66
57,56
66,34
|
1,16
1,09
1,05
1,08
1,09
1,04
|
0,627
0,596
0,631
0,709
0,795
0,836
|
| Т=303 К; <k>=(1,09±0,01)·10-7
, с-1
|
7200
14400
21600
28800
36000
43200
|
0,177
0,147
0,120
0,095
0,065
0,048
|
13,66
28,29
41,46
53,66
68,29
76,59
|
3,12
3,23
3,16
3,06
3,12
2,91
|
1,64
1,85
1,99
2,14
2,56
2,69
|
| Т=313 К; <k>=(3,10±0,04)·10-7
, с-1
|
3600
7200
10800
15780
18000
21600
|
0,170
0,136
0,099
0,055
0,033
0,009
|
17,07
33,66
51,71
73,17
83,90
95,61
|
7,79
7,68
7,87
7,62
7,66
7,28
|
4,17
4,57
5,40
6,68
8,14
11,60
|
| Т=323 К; <k>=(7,65±0,07)·10-7
, с-1
|
1205
2400
3600
4800
6000
7200
|
0,174
0,145
0,116
0,089
0,070
0,041
|
15,12
29,27
43,41
56,59
65,85
80,00
|
20,63
20,04
19,82
19,38
18,04
18,26
|
10,91
11,57
12,68
13,94
14,36
17,92
|
| Т=333 К; <k>=(19,36±0,35)·10-7
, с-1
|
Таблиця 3.18 - Кінетика реакції EtOСН2
СООН (а=0,172 – 0,199 М) з ЕХГ (b=12,48 М) у присутності каталізатора (C2
H5
)4
NBr (m=0,005М)
| t, c |
(а-х), М |
у, % |
kсп
·107
, с-1
|
kсп
·106
, л/моль· с |
17400
34800
52200
69600
87000
104400
|
0,164
0,145
0,125
0,105
0,088
0,071
|
17,59
27,14
37,19
47,24
55,78
64,32
|
0,890
0,740
0,884
0,853
0,850
0,883
|
0,891
0,729
0,714
0,736
0,752
0,791
|
| Т=303 К; <k>=(0,850±0,019)·10-7
, с-1
|
6780
13800
18000
24060
30600
37800
|
0,155
0,133
0,123
0,106
0,090
0,069
|
9,88
22,67
28,49
38,37
47,67
59,88
|
2,01
2,27
2,18
2,20
2,15
2,18
|
1,23
1,49
1,49
1,61
1,70
1,94
|
| Т=313 К; <k>=(2,17±0,03)·10-7
, с-1
|
2700
5400
8100
10860
13560
16200
|
0,164
0,145
0,122
0,104
0,085
0,066
|
10,38
20,77
33,33
43,17
53,55
63,93
|
5,64
5,64
6,04
5,83
5,79
5,79
|
3,25
3,45
4,01
4,17
4,53
5,05
|
| Т=323 К; <k>=(5,79±0,05)·10-7
, с-1
|
1800
3000
4200
5400
6600
7800
|
0,151
0,134
0,102
0,069
0,040
0,016
|
24,12
32,66
48,74
65,33
79,90
91,96
|
19,37
17,37
18,51
19,30
19,31
18,81
|
12,29
10,57
12,75
15,72
19,49
25,90
|
| Т=333 К; <k>=(18,79±0,27)·10-7
, с-1
|
На основі отриманих експериментальних даних були побудовані графіки залежності в координатах lg kсп
від 1/Т.
Рис. 3.12 Залежність ln kсп
від 1/Т реакцій ацидолізу ЕХГ оцтовою кислотою у присутності різних каталізаторів (m=0,005 М) 1 - (C2
H5
)4
NBr; 2 - 4-Me-C5
H4
N; 3 - C6
H5
N(CH3
)2
; 4 - 4-Br-C6
H4
N(CH3
)2
; 5 - 3-NO2
-C6
H4
N(CH3
)

Рис.3.13 Залежність lnkсп
від 1/T реакцій ацидолізу ЕХГ карбоновими кислотами (0,170 – 0,210 М) у присутності Et4
NBr (m=0,005 М) 1 - (СН3
)3
ССООН; 2 - (СН3
)2
СНСООН; 3 - С2
Н5
СН2
СООН; 4 - С2
Н5
ОСН2
СООН; 5 - СН3
СООН
Залежність у координатах ln kсп
від 1/Т як при варіюванні каталізаторів, так і у випадку варіювання замісників в молекулі оцтової кислоти носить прямолінійний характер, що вказує на незмінність механізму реакції в досліджуваному інтервалі температур. При цьому зазначено, що зменшення основних властивостей каталізатору зменшує активаційні параметри реакції (табл. 3.19) в той самий час, коли варіювання структури кислоти практично не впливає на активаційні параметри реакції (табл. 3.20).
Таблиця 3.19 - Активаційні параметри реакції оцтової кислоти (0,197 – 0,210 М) з ЕХГ (12,48 – 12,59 М) для різних каталізаторів (m=0,005 М)
| Каталізатор |
Еа
, кДж/моль |
ln А |
∆Н#
333
, кДж/моль |
-∆S#
333
, Дж/моль•К |
| Et4
NBr |
78,7±1,7 |
20,1 |
73,2 |
95,1 |
| 4-Me-Py |
77,3±2,2 |
19,0 |
71,7 |
104 |
| ДМА |
69,9±0,3 |
15,9 |
64,4 |
130,3 |
| 4-Br-ДМА |
68,3±3,0 |
14,7 |
62,7 |
139 |
| 3-NO2
-ДМА |
55,4±1,2 |
8,83 |
49,8 |
183 |
Таблиця 3.20 - Активаційні параметри реакції карбонових кислот (0,170 – 0,210 М) з ЕХГ (12,55 – 12,62 М) у присутності Et4
NBr (m=0,005 М)
| Кислота |
Еа
, кДж/моль |
ln А |
∆Н#
333
, кДж/моль |
-∆S#
333
, Дж/моль•К |
| СН3
СООН |
78,7±1,7 |
20,1 |
73,2 |
95,2 |
| С2
Н5
ОСН2
СООН |
82,5±2,1 |
21,8 |
76,9 |
80,9 |
| С2
Н5
СН2
СООН |
80,1±0,9 |
21,1 |
74,6 |
87,2 |
| (СН3
)2
СНСООН |
77,8±0,1 |
20,5 |
72,2 |
92,3 |
| (СН3
)3
ССООН |
79,4±1,1 |
21,8 |
73,8 |
81,1 |
Користуючись тим, що активаційні параметри отримані як при варіюванні кислоти, так і при варіюванні каталізатора, представляється можливим побудувати єдину ізокінетичну залежність у координатах ΔН≠
=f(ΔS≠
) для розглянутих вище реакційних серій, яка носить прямолінійний характер з коефіцієнтом кореляції r=0,990, що свідчить про наявність єдиного механізму реакції за даних умов.
Рис.3.14 Залежність ентальпії активації від ентропії активації для реакцій оцтової кислоти та її похідних (0,170 – 0,210 М) з ЕХГ (12,55 – 12,62 М) у присутності різних каталізаторів при Т=333К
Розраховані активаційні параметри при співставленні рівнянь Ареніуса та Ейрінга відповідають таковим для реакцій бімолекулярного нуклеофільного заміщення [22].
Висновки
На основі дослідження каталітичного ацидолізу епіхлоргідрину оцтовою кислотою та деякими її похідними у присутності каталізаторів N,N-диметиланілінів, γ-піколіну та тетраетиламоній броміду в інтервалі концентрацій 0,00125 – 0,005 М при Т = 303 - 333К було встановлено:
1. Реакція має нульовий порядок за карбоновою кислотою та перший за каталізатором.
2. Тетраетиламоній бромід, γ-піколін, N,N-ДМА, 4-Br-N,N-ДМА та 3-NО2
-N,N-ДМА є ефективними каталізаторами ацидолізу ЕХГ.
3. Реакція має низьку чутливість до основності каталізатору.
4. При введенні в молекулу оцтової кислоти електронодонорних замісників спостерігається зменшення швидкості реакції із збільшенням кислотних властивостей карбонової кислоти на відміну від замісників електроноакцепторного характеру, де, навпаки, спостерігається збільшення швидкості реакції із збільшенням кислотних властивостей кислотного реагенту.
5. Знайдені константи реакційної серії ρ1
*
для електронодонорних замісників та ρ2
*
для електроноакцепторних замісників рівняння Тафта вказують на достатньо високу чутливість процесу до зміни замісника електронодонорного характеру в оцтовій кислоті порівняно до зміни замісника електорноакцепторної природи в даних умовах .
6. З ростом температури спостерігається збільшення швидкості реакції.Розраховані активаційні параметри при співставленні рівнянь Ареніуса та Ейрінга відповідають таковим для реакцій бімолекулярного нуклеофільного заміщення.
7. Наявність ізокінетичної залежності свідчить про єдиний механізм реакції як для карбонових кислот з електронодонорними, так і електроноакцепторними замісниками та для каталізаторів, рКа
яких змінюється майже на 15 порядків.
Література
1. Лебедев Н. Н., Гуськов К. А. Кинетика реакции окиси этилена с уксусной и монохлоруксусной кислотами // Кинетика и катализ. – 1963. – Т. 4.-вып. 1. – С. 116-127.
2. Шологон И. М., Клебанов М. С., Алдошин В. А. Катализ реакций эпихлоргидрина с 4-метил-3,4-тетрагидрофталевой кислотой галогенидами тетраелкиламмония // Кинетика и катализ. – 1982. – Т. 13, вып. 4. – С. 841-846.
3. Лебедев Н. Н., Гуськов К. А. Реакционная способность карбоновых кислот в реакции с окисью этилена // Кинетика и катализ. – 1964. – Т. 5, вып. 5. – С. 787-791.
4. Bukowski W. The Solvent Effects in the Reactions of Carboxylic Acids with Oxiranes // Int. J. Chem. Kinet. – 2000. – V. 32, №6. – P. 378-387.
5. Пакен А. М. Эпоксидные соединения и эпоксидные смолы / А. М. Пакен / Л.: Госхимиздат, 1962. - С. 568 – 614.
6. Направление раскрытия α-оксидного кольца в реакции эпихлоргидрина с карбоновыми кислотами при основном катализе. / М. Ф. Сорокин, Л. Г. Шодэ, А. И. Кузьмин, Н. А. Новиков // Кинетика и катализ. – 1967. – Т. 8, вып. 3. – С. 512-519.
7. Mechanism of Acid-catalised Alcoholysis of Epoxides. Part V. Reactions of Substituted (1,2-epoxyethyl)benzenes. / By I. Biggs, N. B. Chapman, A. F. Finch, and V. Wray. Department of Chemistry, The University, Hull HU 6 7RX I. Chem. Soc. (B), 1971; p. 55-65
8. Bukowska A., Bukowski W. Kinetic Study of Addition of Some Carboxylic Acids to 1,2-Epoxy-3-phenoxypropane // Organic Process Research & Development. – 1999. – V. 3. – P. 432-436.
9. Механизм и кинетика основного катализа реакции уксусной кислоты с эпоксидами. / А. К. Гуськов, Сушен Юй, М. Г. Макаров и др.// Кинетика и катализ. – 1994. – Т. 35, №6. – С. 873-877.
10. Справочник химика / Под ред. Никольского В. П. и др. – М.-Л.: Химия, 1971. – Т.2. – 1168 с.
11. Получение оптически прозрачной хлорсодержащей эпоксидной смолы / Л. М. Литвиненко, Р. С. Попова, Т. Н. Соломойченко и др. – Деп. в УкрНИИНТИ, №638. Ук-84. – 1984. – № 8 (154). – 9 с.
12. Fikling M.M., Fisher A., Mann B.R. Hammet substituted constants for electron-withdrawing substituents: dissociation of phenols, anilinium ions and dimethylanilinium ions //Journal of American Chemical Society – 1959. – V. 81. – P. 4226-4230.
13. Свойства органических соединений. Справочник. / Под ред. А.А.Потехина. Л.: Химия. - 1984. - 520 с.
14. Вольский К.П., Хвостов И.В. Методы получения и очистки некоторых четвертичных аммониевых солей, применяемых в полярографии. – М.: НИИТЭхим, 1975. – 8 с.
15. Синтезы органических препаратов. Сб. 1. Под ред. акад. Б. А. Казанского / М.: Изд. иностр. лит., 1949. – С. 412-413.
16. Синтезы органических препаратов. Сб. 2. Под ред. акад. Б. А. Казанского / М.: Изд. иностр. лит., 1949. – С. 607-609.
17. Эммануэль Н. М., Кнорре Д. Г. Курс химической кинетики. – М.: Высшая школа, 1969. – 431 с.
18. Линник Ю. В. Метод наименьших квадратов и основы теории обработки наблюдений. – М.: Физ. Мат. Изд., 1961. – С. 304-343.
19. Вредные вещества в промышленности / под ред. Н. В. Лазарева. – Л.: Госхимиздат, 1954. – Т. 1 – 810 с.
20. Гитис С.С., Глаз А.И., Иванов А.В. Практикум по органической химии. – М.: Высш. шк., 1991. – 303 с.
21. Юрьев Ю.К. Практические работы по органической химии. – Изд-во Московского университета, 1964. – Вып. 1, 2. 3-е издание. – С. 292-293.
22. А. Гордон, Р. Форд Спутник химика. - М.: Мир, 1976. - С. 160.
|