| Вступление
Со времени обнаружения в митохондриях молекул ДНК прошло четверть ве-ка, прежде чем ими заинтересовались не только молекулярные биологи и цито-логи, но и генетики, эволюционисты, а также палеонтологи и криминалисты. Такой широкий интерес спровоцировала работа А.Уилсона из Калифорнийско-го университета. В 1987 г. он опубликовал результаты сравнительного анализа ДНК митохондрий, взятых у 147 представителей разных этносов всех человече-ских рас, заселяющих пять континентов. По типу, местоположению и количес-тву индивидуальных мутаций установили, что все митохондриальные ДНК воз-никли из одной предковой последовательности нуклеотидов путем диверген-ции. В околонаучной прессе вывод этот интерпретировали крайне упрощенно — все человечество произошло от одной женщины, названной митохондриаль-ной Евой (т.к. и дочери и сыновья получают митохондрии только от матери), которая жила в Северо-Восточной Африке около 200 тыс. лет назад. Еще через 10 лет удалось расшифровать фрагмент ДНК митохондрий, выделенный из ос-танков неандертальца, и оценить время существования последнего общего предка человека и неандертальца в 500 тыс. лет назад.
Сегодня митохондриальная генетика человека интенсивно развивается как в популяционном, так и в медицинском аспекте. Установлена связь между рядом тяжелых наследственных заболеваний и дефектами в митохондриальных ДНК. Генетические изменения, ассоциированные со старением организма, наиболее выражены в митохондриях. Что же представляет из себя геном митохондрий, отличающийся у человека и других животных от такового у растений, грибов и простейших и по размеру, и по форме, и по генетической емкости? Какова роль, как работает и как возник митохондриальный геном у разных таксонов в целом и у человека в частности? Об этом и пойдет речь в моем “маленьком и самом скромном” реферате.
У всех эвкариот — будь это малярийный плазмодий, мельчайший одноклето-чный паразит, разрушающий эритроциты человека, или сам человек, гигантская свободноживущая клетка амеба протей, микроскопическая колония дрожжей или гриб, имеющий многокилометровый мицелий, эфемерные насекомые поде-нки или тысячелетние секвойи — у всех генетическая информация содержится не только в хромосомах клеточного ядра, но и в митохондриях — само-воспроизводящихся полуавтономных органеллах клетки, имеющих собствен-ный геном. В то время как ядерный геном представляет собой совокупность линейных молекул ДНК гаплоидного набора хромосом, митохондриальный ге-ном — одну или несколько кольцевых(редко линейных)молекул ДНК (мтДНК). В исключительных случаях эвкариотические клетки не содержат митохондрий, например некоторые паразитирующие в кишечнике анаэробные амебы.
В матриксе митохондрий, кроме ДНК, находятся и собственные рибосомы, по многим характеристикам отличающиеся от эвкариотических рибосом, рас-положенных на мембранах эндоплазматической сети. Однако на рибосомах ми-тохондрий образуется не более 5% от всех белков, входящих в их состав. Бóль-шая часть белков, составляющих структурные и функциональные компоненты митохондрий, кодируется ядерным геномом, синтезируется на рибосомах эндо-плазматической сети и транспортируется по ее каналам к месту сборки. Таким образом, митохондрии — это результат объединенных усилий двух геномов и двух аппаратов транскрипции и трансляции. Некоторые субъединичные ферме-нты дыхательной цепи митохондрий состоят из разных полипептидов, часть ко-торых кодируется ядерным, а часть — митохондриальным геномом. Например, ключевой фермент окислительного фосфорилирования — цитохром-с-оксидаза у дрожжей состоит из трех субъединиц, кодируемых и синтезируемых в мито-хондриях, и четырех, кодируемых в ядре клетки и синтезируемых в цитоплазме. Экспрессией большинства генов митохондрий управляют определенные гены ядер.
Симбиотическая теория происхождения митохондрий
Гипотезу о происхождении митохондрий и растительных пластид из вну-триклеточных бактерий-эндосимбионтов высказал Р.Альтман еще в 1890 г. За век бурного развития биохимии, цитологии, генетики и появившейся полвека назад молекулярной биологии гипотеза переросла в теорию, основанную на бо-льшом фактическом материале. Суть ее такова: с появлением фотосинтезирую-щих бактерий в атмосфере Земли накапливался кислород — побочный продукт их метаболизма. С ростом его концентрации усложнялась жизнь анаэробных ге-теротрофов, и часть из них для получения энергии перешла от бескислородного брожения к окислительному фосфорилированию. Такие аэробные гетеротрофы могли с бóльшим КПД, чем анаэробные бактерии, расщеплять органические ве-щества, образующиеся в результате фотосинтеза. Часть свободно живущих аэ-робов была захвачена анаэробами, но не “переварена”, а сохранена в качестве энергетических станций, митохондрий. Не стоит рассматривать митохондрии как рабов, взятых в плен, чтобы снабжать молекулами АТФ не способные к ды-ханию клетки. Они скорее “существа”, еще в протерозое нашедшие для себя и своего потомства лучшее из убежищ, где можно затрачивать наименьшие уси-лия, не подвергаясь риску быть съеденными.
В пользу симбиотической теории говорят многочисленные факты:
— совпадают размеры и формы митохондрий и свободно живущих аэробных бактерий; те и другие содержат кольцевые молекулы ДНК, не связанные с гистонами (в отличие от линейных ядерных ДНК);
— по нуклеотидным последовательностям рибосомные и транспортные РНК митохондрий отличаются от ядерных, демонстрируя при этом удивительное сходство с аналогичными молекулами некоторых аэробных грамотрицательных эубактерий;
— митохондриальные РНК-полимеразы, хотя и кодируются в ядре клетки, ингибируются рифампицином, как и бактериальные, а эвкариотические РНК-полимеразы нечувствительны к этому антибиотику;
— белковый синтез в митохондриях и бактериях подавляется одними и теми же антибиотиками, не влияющими на рибосомы эвкариот;
— липидный состав внутренней мембраны митохондрий и бактериальной плазмалеммы сходен, но сильно отличается от такового наружной мембраны митохондрий, гомологичной другим мембранам эвкариотических клеток;
— кристы, образуемые внутренней митохондриальной мембраной, являются эволюционными аналогами мезосомных мембран многих прокариот;
— до сих пор сохранились организмы, имитирующие промежуточные формы на пути к образованию митохондрий из бактерий (примитивная амеба Pelomyxa
не имеет митохондрий, но всегда содержит эндосимбиотические бактерии).
Существует представление, что разные царства эвкариот имели разных предков и эндосимбиоз бактерий возникал на разных этапах эволюции живых организмов. Об этом же говорят отличия в строении митохондриальных гено-мов простейших, грибов, растений и высших животных. Но во всех случаях ос-новная часть генов из промитохондрий попала в ядро, возможно, с помощью мобильных генетических элементов. При включении части генома одного из симбионтов в геном другого интеграция симбионтов становится необратимой. Новый геном может создавать метаболические пути, приводящие к образова-нию полезных продуктов, которые не могут быть синтезированы ни одним из партнеров по отдельности. Так, синтез стероидных гормонов клетками коры надпочечников представляет собой сложную цепь реакций, часть которых происходит в митохондриях, а часть — в эндоплазматической сети. Захватив гены промитохондрий, ядро получило возможность надежно контролировать функции симбионта. В ядре кодируются все белки и синтез липидов наружной мембраны митохондрий, большинство белков матрикса и внутренней мембраны органелл. Самое главное, что ядро кодирует ферменты репликации, транскрип-ции и трансляции мтДНК, контролируя тем самым рост и размножение мито-хондрий. Скорость роста партнеров по симбиозу должна быть приблизительно одинаковой. Если хозяин будет расти быстрее, то с каждым его поколением число симбионтов, приходящихся на одну особь, будет уменьшаться, и, в конце концов, появятся потомки, не имеющие митохондрий. Мы знаем, что в каждой клетке организма, размножающегося половым путем, содержится много мито-хондрий, реплицирующих свои ДНК в промежутке между делениями хозяина. Это служит гарантией того, что каждая из дочерних клеток получит по крайней мере одну копию генома митохондрии.
Роль клеточного ядра в биогенезе митохондрий
У мутантных дрожжей определенного типа имеется обширная делеция в митохондриальной ДНК, что ведет к полному прекращению белкового синтеза в митохондриях; в результате эти органеллы не способны выполнять, свою функцию. Так как при росте на среде с низким содержанием глюкозы такие мутанты образуют мелкие колонии, их называют цитоплазматическими му
тантами
petite
.
Хотя у мутантов petite нет митохондриального синтеза белков и поэтому нормальных митохондрий не образуется, тем не менее такие мутанты содержат промитохондрии,
которые в известной мере сходны с обычными митохондриями, имеют нормальную наружную мембрану и внутреннюю мeмбрану со слабо развитыми кристами. В промитохондриях имеются многие ферменты, кодируемые ядерными генами и синтезируемые на рибосомах цитоплазмы, в том числе ДНК- и РНК-полимеразы, все ферменты цикла лимонной кислоты и многие белки, входящие в состав внутренней мембраны. Это наглядно демонстрирует преобладающую роль ядерного генома в биогенезе митохондрий.
Интересно отметить, что, хотя утраченные фрагменты ДНК составляют от 20 до более чем 99,9% митохондриального генома, общее количество митохондриальной ДНК у мутантов petite всегда остается на том же уровне, что и у дикого типа. Это обусловлено еще мало изученным процессом aмплификации ДНК, в результате которого образуется молекула ДНК, состоящая из тандемных повторов одного и того же участка и равная по величине нормальной молекуле. Например, митохондриальная ДНК мутанта petite, сохранившая 50% нуклеотидной последовательности ДНК дикого типа, будет состоять из двух повторов, тогда как молекула, сохранившая только 0,1%
генома дикого типа, будет построена из 1000 копий оставшегося фрагмента. Таким образом, мутанты petite могут быть использованы для получения в большом количестве определенных участков митохондриальной ДНК, которые, можно сказать, клонируются самой природой.
Хотя биогенез органелл контролируется главным образом ядерными генами, сами органеллы тоже, судя по некоторым данным, оказывают какое-то регулирующее влияние по принципу обратной связи; во всяком случае так обстоит дело с митохондриями. Если блокировать синтез белка в митохондриях интактных клеток, то в цитоплазме начинают в избытке образовываться ферменты участвующие в митохондриальном синтезе ДНК, РНК и белков, как будто клетка пытается преодолеть воздействие блокирующего агента. Но, хотя существование какого-то сигнала со стороны митохондрий и не вызывает сомнений, природа его до сих пор не известна.
По ряду причин механизмы биогенеза митохондрий изучают сейчас в большинстве случаев на культурах Saccharomyces
carlsbergensis
(пивные дрожжи и S
.
cerevisiae
(пекарские дрожжи). Во-первых, при росте на глюкозе эти дрожжи обнаруживают уникальную способность существовать только за счет гликолиза, т.е. обходиться без функции митохондрий. Это дает возможность изучать мутации в митохондриальной и ядерной ДНК, препятствующие развитию этих органелл. Такие мутации летальны почти у всех других организмов. Во-вторых, дрожжи - простые одноклеточные эукариоты- легко культивировать и подвергать биохимическому исследованию. И наконец, дрожжи могут размножаться как в гаплоидной, так и в диплоидной фазе, обычно бесполым способом-почкованием (асимметричный митоз). Но у дрожжей встречается и половой процесс: время от времени две гаплоидные клетки сливаются, образуя диплоидную зиготу, которая затем либо делится путем митоза, либо претерпевает мейоз и снова дает гаплоидные клетки. Контролируя в ходе эксперимента чередование бесполого и полового раз-множения, можно многое узнать о генах, ответственных за функцию митохондрий. С помощью этих методов можно, в частности, выяснить, локализованы ли такие гены в ядерной ДНК или в митохондриальной, так как мутации митохондриальных генов не наследуются по законам Менделя, которым подчиняется наследование ядерных генов.
Транспортные системы митохондрий
Большая часть белков, содержащихся в митохондриях и хлоропластах импор-тируется в эти органеллы из цитозоля. В связи с этим возникают два вопроса: как клетка направляет белки к надлежащей органелле и каким образом эти белки проникают в нее?
Частичный ответ был получен при изучении транспорта в строму хлоропласта малой субъединицы (S) фермента рибулозо-1,5-бисфосфат-карбокси
лазы.
Если мРНК, выделенную из цитоплазмы одноклеточной водоросли Chlamydomonas
или из листьев гороха, ввести в качестве матрицы в белоксинтезирующую систему in vitro, то один из многих образующихся белков будет связываться специфическим анти-S-антителом. S-белок, синтезируемый in vitro, называют пpo-S, так как он больше обычного S-белка примерно на 50 аминокислотных остатков. При инкубации белка пpo-S с интактными хлоропластами он проникает в органеллы и превращается там под действием пептидазы в S-белок. Затем S-белок связывается с большой субъединицей рибулозо-1,5-бисфосфат-карбоксилазы, синтезируемой на рибосомах хлоропласта, и образует с нею в строме хлоропласта активный фермент.
Механизм переноса S-белка неизвестен. Полагают, что пpo-S связывается с белком-рецептором, находящимся на наружной мембране хлоропласта или в месте контакта наружной и внутренней мембран, а затем переносится в строму через трансмембранные каналы в результате процесса, требующего затраты энергии.
Сходным образом осуществляется транспорт белков внутрь митохондрий. Если очищенные митохондрии дрожжей инкубировать с клеточным экстрактом, содержащим только что синтезированные радиоактивные дрожжевые белки, то можно наблюдать, что митохондриальные белки, кодируемые ядерным геномом, отделяются от немитохондриальных белков цитоплазмы и избирательно включаются в митохондрии-так же, как это происходит в интактной клетке. При этом белки наружной и внутренней мембран, матрикса и межмембранного пространства находят свой путь к соответствующему компартменту митохондрии.
Многие из вновь синтезированных белков, предназначенных для внутренней мембраны, матрикса и межмембранного пространства, имеют на своем N-конце лидерный пептид, который во время транспортировки отщепляется специфической протеазой, находящейся в матриксе. Для переноса белков в эти три митохондриальных компартмента необходима энергия электрохимического протонного градиента, создаваемого на внутренней мембране. Механизм переноса белков для наружной мембраны иной: в этом случае не требуется ни затрат энергии, ни протеолитического расщепления более длинного белка-предшественника. Эти и другие наблюдения позволяют думать, что все четыре группы митохондриальных белков транспортируются в органеллу с помощью следующего механизма: предполагается, что все белки, кроме тех, которые предназначены для наружной мембраны, включаются во внутреннюю мембрану в результате процесса, требующего затраты энергии и происходящего в местах контакта наружной и внутренней мембран. По-видимому, после этого первоначального включения белка в мембрану он подвергается протеолитическому расщеплению, которое приводит к изменению его конформации; в зависимости от того, как изменится конформация, белок либо закрепляется в мембране, либо «выталкивается» в матрикс или в межмембранное пространство.
Перенос белков через мембраны митохондрий и хлоропластов в принципе аналогичен переносу их через мембраны эндоплазматического ретикулума. Однако здесь есть несколько важных отличий. Во-первых, при транспорте в матрикс или строму белок проходит как через наружную, так и через внутреннюю мембрану органеллы, тогда как при переносе в просвет эндоплазматического ретикулума молекулы проходят только через одну мембрану. Кроме того, перенос белков в ретикулум осуществляется с помощью механизма направленного выведения
(vectorial discharge)-он начинается тогда, когда белок еще не полностью сошел с рибосомы (котрансляционный импорт),
а перенос в митохондрии и хлоропласты происходит уже после того, как синтез белковой молекулы будет полностью завершен (посттрансляционный импорт).
Несмотря на эти различия, и в том и в другом случае клетка синтезирует белки-предшественники, содержащие сигнальную последовательность, которая определяет, к какой мембране направится данный белок. По-видимому, во многих случаях эта последовательность отщепляется от молекулы-предшественника после завершения транспортного процесса. Однако некоторые белки сразу синтезируются в окончательном виде. Полагают, что в таких случаях сигнальная последовательность заключена в полипептидной цепи готового белка. Сигнальные последовательности еще плохо изучены, но, вероятно, должно быть несколько типов таких последовательностей, каждый из которых определяет перенос белковой молекулы в определенную область клетки. Например, в растительной клетке некоторые из белков, синтез которых начинается в цитозоле, транспортируются затем в митохондрии, другие - в хлоропласты, третьи - в пероксисомы, четвертые - в эндоплазматический ретикулум. Сложные процессы, приводящие к правильному внутриклеточному распределению белков, только сейчас становятся понятными.
Помимо нуклеиновых кислот и белков для построения новых митохондрий нужны липиды. В отличие от хлоропластов митохондрии получают бóльшую часть своих липидов извне. В животных клетках фосфолипиды, синтезированные в эндоплазматическом ретикулуме, транспортируются к наружной мембране митохондрий с помощью особых белков, а затем включаются во внутреннюю мембрану; как полагают, это происходит в месте контакта двух мембран. Основная реакция биосинтеза липидов, катализируемая самими митохондриями, - это превращение фосфатидной кислоты в фосфолипид кардиолипин, который содержится главным образом во внутренней митохондриальной мембране и составляет около 20% всех ее липидов.
Размеры и форма митохондриальных геномов
К настоящему времени прочитано более 100 разных геномов митохондрий. На-бор и количество их генов в митохондриальных ДНК, для которых полностью определена последовательность нуклеотидов, сильно различаются у разных ви-дов животных, растений, грибов и простейших. Наибольшее количество генов обнаружено в митохондриальном геноме жгутикового простейшего Rectinomo-nas americana
— 97 генов, включая все кодирующие белок гены, найденные в мтДНК других организмов. У большинства высших животных геном митохон-дрий содержит 37 генов: 13 для белков дыхательной цепи, 22 для тРНК и два для рРНК (для большой субъединицы рибосом 16S рРНК и для малой 12S рРНК). У растений и простейших, в отличие от животных и большинства гри-бов, в митохондриальном геноме закодированы и некоторые белки, входящие в состав рибосом этих органелл. Ключевые ферменты матричного полинуклеоти-дного синтеза, такие как ДНК-полимераза (осуществляющая репликацию мито-хондриальной ДНК) и РНК-полимераза (транскрибирующая геном митохон-дрий), зашифрованы в ядре и синтезируются на рибосомах цитоплазмы. Этот факт указывает на относительность автономии митохондрий в сложной иерар-хии эвкариотической клетки.
Геномы митохондрий разных видов отличаются не только по набору ге-нов, порядку их расположения и экспрессии, но по размеру и форме ДНК. По-давляющее большинство описанных сегодня митохондриальных геномов пред-ставляет собой кольцевые суперспирализованные двуцепочечные молекулы ДНК. У некоторых растений наряду с кольцевыми формами имеются и линей-ные, а у некоторых простейших, например инфузорий, в митохондриях обнару-жены только линейные ДНК.
Как правило, в каждой митохондрии содержится несколько копий ее ге-нома. Так, в клетках печени человека около 2 тыс. митохондрий, и в каждой из них — по 10 одинаковых геномов. В фибробластах мыши 500 митохондрий, со-держащих по два генома, а в клетках дрожжей S.cerevisiae
— до 22 митохон-дрий, имеющих по четыре генома.
 Митохондриальный геном растений, как правило, состоит из нескольких молекул разного размера. Одна из них, “основная хромосома”, содержит боль-шую часть генов, а кольцевые формы меньшей длины, находящиеся в динами-ческом равновесии как между собой, так и с основной хромосомой, образуются в результате внутри- и межмолекулярной рекомбинации благодаря наличию по-вторенных последовательностей (рис.1). Митохондриальный геном растений, как правило, состоит из нескольких молекул разного размера. Одна из них, “основная хромосома”, содержит боль-шую часть генов, а кольцевые формы меньшей длины, находящиеся в динами-ческом равновесии как между собой, так и с основной хромосомой, образуются в результате внутри- и межмолекулярной рекомбинации благодаря наличию по-вторенных последовательностей (рис.1).
Рис 1.
Схема образования кольцевых молекул ДНК разного размера в митохондриях растений. Рекомбинация происходит по повторенным участкам (обозначены синим цветом).
В митохондриях большинства организмов (кроме высших животных) часть кольцевых молекул ДНК присутствует в виде олигоме-ров, которые можно разделить на три класса: линейные; кольцевые, имеющие контурную длину, кратную длине мономерных колец; цепные, катенаны, состо-ящие из топологически связанных, т.е. продетых друг в друга, мономерных ко-лец (рис.2). Так, в единственной митохондрии простейших из отряда кинето-пластид, включающего эндопаразита человека — трипаносому, содержатся ты-сячи кольцевых молекул ДНК. У Trypanosoma brucei
имеются два типа моле-кул: 45 одинаковых максиколец, каждое из которых состоит из 21 тыс. пар ну-клеотидов, и 5.5 тыс. идентичных друг другу миниколец по 1000 пар нуклео-тидов. Все они, соединяясь в катенаны, образуют переплетенную сеть, которая вместе с белками формирует структуру, называемую кинетопластом.
 Рис 2.
Схема образования линейных (А), кольцевых (Б), цепных (В) олигомеров мтДНК. ori — район начала репликации ДНК. Рис 2.
Схема образования линейных (А), кольцевых (Б), цепных (В) олигомеров мтДНК. ori — район начала репликации ДНК.
Размер генома митохондрий разных организмов колеблется от менее 6 тыс. пар нуклеотидов у малярийного плазмодия (в нем, помимо двух генов рРНК, содержится только три гена, кодирующих белки) до сотен тысяч пар ну-клеотидов у наземных растений (например, у Arabidopsis thaliana
из семейства крестоцветных 366924 пар нуклеотидов). При этом 7—8-кратные различия в ра-змерах мтДНК высших растений обнаруживаются даже в пределах одного се-мейства. Длина мтДНК позвоночных животных отличается незначительно: у человека — 16569 пар нуклеотидов, у свиньи — 16350, у дельфина — 16330, у шпорцевой лягушки Xenopus laevis
— 17533, у карпа — 16400. Эти геномы схо-дны также и по локализации генов, большинство которых располагаются встык; в ряде случаев они даже перекрываются, обычно на один нуклеотид, так что по-следний нуклеотид одного гена оказывается первым в следующем. В отличие от позвоночных, у растений, грибов и простейших мтДНК содержат до 80% не-кодирующих последовательностей. У разных видов порядок генов в геномах митохондрий отличается.
Высокая концентрация активных форм кислорода в митохондриях и сла-бая система репарации увеличивают частоту мутаций мтДНК по сравнению с ядерной на порядок. Радикалы кислорода служат причиной специфических за-мен Ц®Т (дезаминирование цитозина) и Г®Т (окислительное повреждение гуанина), вследствие чего, возможно, мтДНК богаты АТ-парами. Кроме того, все мтДНК обладают интересным свойством — они не метилируются, в отли-чие от ядерных и прокариотических ДНК. Известно, что метилирование (време-нная химическая модификация нуклеотидной последовательности без наруше-ния кодирующей функции ДНК) — один из механизмов программируемой инактивации генов.
Размеры и строение молекул ДНК в органеллах
| Вид
|
Структура
|
Масса, млн.
дальтон
|
Примечания
|
| Мит
охон
дриа
льн
ая
Д
Н
К
|
Животные
|
Кольцевая
|
9-12
|
У каждого отдельного вида все молекулы одного размера
|
| Высшие ра
стения
|
Кольцевая
|
Варьирует
|
У всех изученных видов имеются разные по величине кольцевые ДНК, в которых общее содержание генетической информации соответ-ствует массе от 300 до 1000 млн. дальтон в зависимости от вида
|
| Грибы:
Saccharomyces
Kluyveromyces
Простейшие
Plasmodium
Paramecium
|
Кольцевая
Кольцевая
Кольцевая
Линейная
|
50
22
18
27
|
|
| Д
Н
К
Хлор
опла
стов
|
Водоросли
Chlamydomonas
Euglena
|
Кольцевая
Кольцевая
|
120
90
|
|
| Высшие
растения
|
Кольцевая
|
85-97
|
У каждого отдельного вида найдены молекулы только одного
размера
|
Относительное количество ДНК органелл в некоторых клетках и тканях
| Организм
|
Ткань или
тип клеток
|
Число мол-л ДНК/органел-
лу
|
Число орга-
нелл в
клетке
|
Доля ДНК орга-нелл во всей
ДНК клетки, %
|
| Мит
охон
дриа
льн
ая
Д
Н
К
|
Крыса
|
Печень
|
5-10
|
1000
|
1
|
| Мышь
|
Клетки линии L
|
5-10
|
100
|
<1
|
| Лягушка
|
Яйцеклетка
|
5-10
|
107
|
99
|
| Дрожжи
|
Вегетативные диплоидные клетки
|
2-50
|
2-50
|
15
|
| Д
Н
К
Хлор
опла
стов
|
Chlamydomonas
|
Вегетативные диплоидные клетки
|
80
|
2
|
7
|
| Кукуруза
|
Листья
|
20-40
|
20-40
|
15
|
Функционирование митохондриального генома
Что же особенного в механизмах репликации и транскрипции ДНК митохондрий млекопитающих?
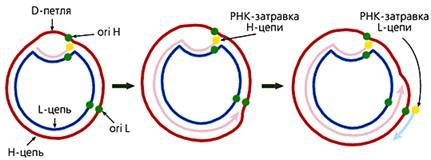 У большинства животных комплементарные цепи в мтДНК значительно различаются по удельной плотности, поскольку содержат неодинаковое количе-ство “тяжелых” пуриновых и “легких” пиримидиновых нуклеотидов. Так они и называются — H (heavy — тяжелая) и L (light — легкая) цепь. В начале репли-кации молекулы мтДНК образуется так называемая D-петля (от англ. Displace-ment loop — петля смещения). Эта структура, видимая в электронный микро-скоп, состоит из двуцепочечного и одноцепочечного (отодвинутой части Н-цепи) участков. Двуцепочечный участок формируется частью L-цепи и компле-ментарным ей вновь синтезированным фрагментом ДНК длиной 450—650 (в зависимости от вида организма) нуклеотидов, имеющим на 5'-конце рибонук-леотидную затравку, которая соответствует точке начала синтеза Н-цепи (oriH). Синтез L-цепи начинается лишь тогда, когда дочерняя Н-цепь доходит до точки ori L. Это обусловлено тем, что область инициации репликации L-цепи доступ-на для ферментов синтеза ДНК лишь в одноцепочечном состоянии, а следовате-льно, только в расплетенной двойной спирали при синтезе Н-цепи. Таким обра-зом, дочерние цепи мтДНК синтезируются непрерывно и асинхронно (рис.3). У большинства животных комплементарные цепи в мтДНК значительно различаются по удельной плотности, поскольку содержат неодинаковое количе-ство “тяжелых” пуриновых и “легких” пиримидиновых нуклеотидов. Так они и называются — H (heavy — тяжелая) и L (light — легкая) цепь. В начале репли-кации молекулы мтДНК образуется так называемая D-петля (от англ. Displace-ment loop — петля смещения). Эта структура, видимая в электронный микро-скоп, состоит из двуцепочечного и одноцепочечного (отодвинутой части Н-цепи) участков. Двуцепочечный участок формируется частью L-цепи и компле-ментарным ей вновь синтезированным фрагментом ДНК длиной 450—650 (в зависимости от вида организма) нуклеотидов, имеющим на 5'-конце рибонук-леотидную затравку, которая соответствует точке начала синтеза Н-цепи (oriH). Синтез L-цепи начинается лишь тогда, когда дочерняя Н-цепь доходит до точки ori L. Это обусловлено тем, что область инициации репликации L-цепи доступ-на для ферментов синтеза ДНК лишь в одноцепочечном состоянии, а следовате-льно, только в расплетенной двойной спирали при синтезе Н-цепи. Таким обра-зом, дочерние цепи мтДНК синтезируются непрерывно и асинхронно (рис.3).
Рис 3.
Схема репликации мтДНК млекопитающих. Сначала формируется D-петля, затем синтезируется дочерняя Н-цепь, потом начинается синтез дочерней L-цепи.
 В митохондриях общее число молекул с D-петлей значительно превыша-ет число полностью реплицирующихся молекул. Обусловлено это тем, что у D-петли есть дополнительные функции — прикрепление мтДНК к внутренней ме-мбране и инициация транскрипции, поскольку в этом районе локализованы промоторы транскрипции обеих цепей ДНК. В отличие от большинства эв-кариотических генов, которые транскрибируются независимо друг от друга, ка-ждая из цепей мтДНК млекопитающих переписывается с образованием одной молекулы РНК, начинающейся в районе ori H. Помимо этих двух длинных мо-лекул РНК, комплементарных Н- и L-цепям, формируются и более короткие участки Н-цепи, которые начинаются в той же точке и заканчиваются на 3'-кон-це гена 16S рРНК (рис.4). Таких коротких транскриптов в 10 раз больше, чем длинных. В результате созревания (процессинга) из них образуются 12S рРНК и 16S рРНК, участвующие в формировании митохондриальных рибосом, а так-же фенилаланиновая и валиновая тРНК. Из длинных транскриптов вырезаются остальные тРНК и образуются транслируемые мРНК, к 3'-концам которых при-соединяются полиадениловые последовательности. 5'-концы этих мРНК не кэ-пируются, что необычно для эвкариот. Сплайсинга (сращивания) не происхо-дит, поскольку ни один из митохондриальных генов млекопитающих не содер-жит интронов. В митохондриях общее число молекул с D-петлей значительно превыша-ет число полностью реплицирующихся молекул. Обусловлено это тем, что у D-петли есть дополнительные функции — прикрепление мтДНК к внутренней ме-мбране и инициация транскрипции, поскольку в этом районе локализованы промоторы транскрипции обеих цепей ДНК. В отличие от большинства эв-кариотических генов, которые транскрибируются независимо друг от друга, ка-ждая из цепей мтДНК млекопитающих переписывается с образованием одной молекулы РНК, начинающейся в районе ori H. Помимо этих двух длинных мо-лекул РНК, комплементарных Н- и L-цепям, формируются и более короткие участки Н-цепи, которые начинаются в той же точке и заканчиваются на 3'-кон-це гена 16S рРНК (рис.4). Таких коротких транскриптов в 10 раз больше, чем длинных. В результате созревания (процессинга) из них образуются 12S рРНК и 16S рРНК, участвующие в формировании митохондриальных рибосом, а так-же фенилаланиновая и валиновая тРНК. Из длинных транскриптов вырезаются остальные тРНК и образуются транслируемые мРНК, к 3'-концам которых при-соединяются полиадениловые последовательности. 5'-концы этих мРНК не кэ-пируются, что необычно для эвкариот. Сплайсинга (сращивания) не происхо-дит, поскольку ни один из митохондриальных генов млекопитающих не содер-жит интронов.
| ND1—
ND6,
ND4
L — гены субъединиц Н
AД-
H-дегидрогеназного комплекса; СО
I—
COIII — гены субъединиц цитохром-с-оксидазы;
ATP6,
ATP8 — гены субъединиц
AТФ-синтетазы
Cyt
b — ген цитохрома
b.
|
Рис 4.
Транскрипция мтДНК человека, содержащей 37 генов. Все транскрипты начинают синтезироваться в районе ori H. Рибосомные РНК вырезаются из длинного и короткого транскриптов Н-цепи. тРНК и мРНК образуются в результате процессинга из транскриптов обеих цепей ДНК. Гены тРНК обозначены светло-зеленым цветом.
Хотите узнать какие еще сюрпризы способен преподнести митохон-дриальный геном? Отлично! Читаем дальше!..
 Несмотря на то, что в геномах митохондрий млекопитающих и дрожжей содержится приблизительно одинаковое количество генов, размеры дрожжево-го генома в 4-5 раз больше — около 80 тыс. пар нуклеотидов. Хотя кодирую-щие последовательности мтДНК дрожжей высоко гомологичны соответствую-щим последовательностям у человека, дрожжевые мРНК дополнительно имеют 5'-лидерную и 3'-некодирующую области, как и большинство ядерных мРНК. Ряд генов содержит еще и интроны. Так, в гене box, кодирующем цитохром-оксидазу b, имеется два интрона. Из первичного РНК-транскрипта автокатали-тически (без участия каких-либо белков) вырезается копия большей части пер-вого интрона. Оставшаяся РНК служит матрицей для образования фермента ма-туразы, участвующей в сплайсинге. Часть ее аминокислотной последовательно-сти закодирована в оставшихся копиях интронов. Матураза вырезает их, разру-шая свою собственную мРНК, копии экзонов сшиваются, и образуется мРНК для цитохромоксидазы b (рис.5). Открытие такого феномена заставило пере-смотреть представление об интронах, как о “ничего не кодирующих последова-тельностях”. Несмотря на то, что в геномах митохондрий млекопитающих и дрожжей содержится приблизительно одинаковое количество генов, размеры дрожжево-го генома в 4-5 раз больше — около 80 тыс. пар нуклеотидов. Хотя кодирую-щие последовательности мтДНК дрожжей высоко гомологичны соответствую-щим последовательностям у человека, дрожжевые мРНК дополнительно имеют 5'-лидерную и 3'-некодирующую области, как и большинство ядерных мРНК. Ряд генов содержит еще и интроны. Так, в гене box, кодирующем цитохром-оксидазу b, имеется два интрона. Из первичного РНК-транскрипта автокатали-тически (без участия каких-либо белков) вырезается копия большей части пер-вого интрона. Оставшаяся РНК служит матрицей для образования фермента ма-туразы, участвующей в сплайсинге. Часть ее аминокислотной последовательно-сти закодирована в оставшихся копиях интронов. Матураза вырезает их, разру-шая свою собственную мРНК, копии экзонов сшиваются, и образуется мРНК для цитохромоксидазы b (рис.5). Открытие такого феномена заставило пере-смотреть представление об интронах, как о “ничего не кодирующих последова-тельностях”.
Рис 5.
Процессинг (созревание) мРНК цитохромоксидазы b в митохондриях дрожжей. На первом этапе сплайсинга образуется мРНК, по которой синтезируется матураза, необходимая для второго этапа сплайсинга.
При изучении экспрессии митохон-дриальных генов Trypanosoma brucei
обнаружилось удивительное отклонение от одной из основных аксиом молекулярной биологии, гласящей, что после-довательность нуклеотидов в мРНК в точности соответствует таковой в коди-рующих участках ДНК. Оказалось, мРНК одной из субъединиц цитохром-с-оксидазы редактируется, т.е. после транскрипции изменяется ее первичная структура — вставляется четыре урацила. В результате образуется новая мРНК, служащая матрицей для синтеза дополнительной субъединицы фермента, пос-ледовательность аминокислот в которой не имеет ничего общего с последова- тельностью, кодируемой нередактированной мРНК (см. таблицу). тельностью, кодируемой нередактированной мРНК (см. таблицу).
Происходит это за счет сдвига рамки считыва-ния на число нуклеотидов, не кратное размеру триплета (в данном случае на четыре). Новая белковая субъединица, необходимая для работы фермента, об-разуется в митохондриях паразита только тогда, когда он попадает в организм холоднокровной мухи и нуждается в окислительном фосфорилировании для получения большого количества молекул АТФ. Если трипаносома живет в ор-ганизме теплокровных млекопитающих, ей достаточно АТФ, образующейся в процессе гликолиза. Впервые обнаруженное в митохондриях трипаносомы ре-дактирование РНК широко распространено в хлоропластах и митохондриях вы-сших растений. Найдено оно и в соматических клетках млекопитающих, напри-мер, в кишечном эпителии человека редактируется мРНК гена аполипопротеина.
Наибольший сюрприз ученым митохондрии преподнесли в 1979 г. До то-го времени считалось, что генетический код универсален и одни и те же трип-леты кодируют одинаковые аминокислоты у бактерий, вирусов, грибов, расте-ний и животных. Английский исследователь Беррел сопоставил структуру од-ного из митохондриальных генов теленка с последовательностью аминокислот в кодируемой этим геном субъединице цитохромоксидазы. Оказалось, что гене-тический код митохондрий крупного рогатого скота (как и человека) не просто отличается от универсального, он “идеален”, т.е. подчиняется следующему пра-вилу: “если два кодона имеют два одинаковых нуклеотида, а третьи нуклеоти-ды принадлежат к одному классу (пуриновых — А, Г, или пиримидиновых — У, Ц), то они кодируют одну и ту же аминокислоту”. В универсальном коде есть два исключения из этого правила: триплет АУА кодирует изолейцин, а кодон АУГ — метионин, в то время как в идеальном коде митохондрий оба эти трип-лета кодируют метионин; триплет УГГ кодирует лишь триптофан, а триплет УГА — стоп-кодон. В универсальном коде оба отклонения касаются прин-ципиальных моментов синтеза белка: кодон АУГ — инициирующий, а стоп-кодон УГА останавливает синтез полипептида. Идеальный код присущ не всем описанным митохондриям, но ни у одной из них нет универсального кода. Мож-но сказать, что митохондрии говорят на разных языках, но никогда — на языке ядра.
Различия между “универсальным” генетическим кодом и двумя митохондриальными кодами
| Кодон
|
Митохондриальный
код млекопитающих
|
Митохондриальный
код дрожжей
|
“
Универсальный
”
код
|
| UGA
|
Trp
|
Trp
|
Stop
|
| AUA
|
Met
|
Met
|
Ile
|
| CUA
|
Leu
|
Thr
|
Leu
|
| AGA
AGG
|
Cmon
|
Arg
|
Arg
|
Как уже говорилось, в митохондриальном геноме позвоночных есть 22 ге-на тРНК. Каким же образом такой неполный набор обслуживает все 60 кодонов для аминокислот (в идеальном коде из 64 триплетов четыре стоп-кодона, в уни-версальном — три)? Дело в том, что при синтезе белка в митохондриях упроще-ны кодон-антикодонные взаимодействия — для узнавания используется два из трех нуклеотидов антикодона. Таким образом, одна тРНК узнает все четыре представителя кодонового семейства, отличающиеся только третьим нуклеоти-дом. Например, лейциновая тРНК с антикодоном ГАУ встает на рибосоме на-против кодонов ЦУУ, ЦУЦ, ЦУА и ЦУГ, обеспечивая безошибочное включе-ние лейцина в полипептидную цепь. Два других лейциновых кодона УУА и УУГ узнаются тРНК с антикодоном ААУ. В целом, восемь разных молекул тРНК узнают восемь семейств по четыре кодона в каждом, и 14 тРНК узнают разные пары кодонов, каждая из которых шифрует одну аминокислоту.
Важно, что ферменты аминоацил-тРНК-синтетазы, ответственные за при-соединение аминокислот к соответствующим тРНК митохондрий, кодируются в ядре клетки и синтезируются на рибосомах эндоплазматической сети. Таким образом, у позвоночных животных все белковые компоненты митохондриаль-ного синтеза полипептидов зашифрованы в ядре. При этом синтез белков в ми-тохондриях не подавляется циклогексимидом, блокирующим работу эвкариоти-ческих рибосом, но чувствителен к антибиотикам эритромицину и хлорамфени-колу, ингибирующим белковый синтез в бактериях. Этот факт служит одним из аргументов в пользу происхождения митохондрий из аэробных бактерий при симбиотическом образовании эвкариотических клеток.
Значение наличия собственной генетической системы для митохондрий
Почему митохондриям необходима собственная генетическая система, тогда как другие органеллы, например пероксисомы и лизосомы ее не имеют? Этот вопрос совсем не тривиален, так как поддержание отдельной генетической сис-темы дорого обходится клетке, если учесть необходимое количество дополни-тельных генов в ядерном геноме. Здесь должны быть закодированы рибосом-ные белки, аминоацил-тРНК-синтетазы, ДНК- и РНК-полимеразы, ферменты процессинга и модификации РНК и т. д. Большинство изученных белков из митохондрий отличаются по аминокислотной последовательности от своих аналогов из других частей клетки, и есть основание полагать, что в этих органе-ллах очень мало таких белков, которые могли бы встретиться еще где-нибудь. Это означает, что только для поддержания генетической системы митохондрий в ядерном геноме должно быть несколько десятков дополнительных генов.При-чины такого “расточительства” неясны, и надежда на то, что разгадка будет найдена в нуклеотидной последовательности митохондриальной ДНК, не опра-вдалась. Трудно представить себе, почему образующиеся в митохондриях бел-ки должны непременно синтезироваться именно там, а не в цитозоле.
Обычно существование генетической системы в энергетических органеллах объясняют тем, что некоторые из синтезируемых внутри органеллы белков слишком гидрофобны, чтобы пройти сквозь митохондриальную мембрану из-вне. Однако изучение АТР-синтетазного комплекса показало, что такое объясне-ние неправдоподобно. Хотя отдельные белковые субъединицы АТР-синтетазы весьма консервативны в ходе эволюции, места их синтеза изменяются. В хлоропластах несколько довольно гидрофильных белков, в том числе четыре из пяти субъединиц F1
-ATPазной части комплекса, образуются на рибосомах внутри органеллы. Напротив, у гриба Neurospora
и в животных клетках весьма гидрофобный компонент (субъединица 9) мембранной части АТРазы синтези-руется на рибосомах цитоплазмы и лишь после этого переходит в органеллу. Различную локализацию генов, кодирующих субъединицы функционально эквивалентных белков у разных организмов, трудно объяснить с помощью какой бы то ни было гипотезы, постулирующей определенные эволюционные преимущества современных генетических систем митохондрий и хлоропластов.
Учитывая все вышесказанное, остается только предположить, что генетическая система митохондрий представляет собой эволюционный тупик. В рамках эндо-симбиотической гипотезы это означает, что процесс переноса генов эндосимбионта в ядерный геном хозяина прекратился раньше, чем был полностью завершен.
Цитоплазматическая наследственность
Последствия цитоплазматической передачи генов для некоторых животных, в том числе и для человека, более серьезны, нежели для дрожжей. Две сливающиеся гаплоидные дрожжевые клетки имеют одинаковую величину и вносят в образующуюся зиготу одинаковое количество митохондриальной ДНК. Таким образом, у дрожжей митохондриальный геном наследуется от обоих родителей, которые вносят равный вклад в генофонд потомства (хотя, спустя несколько генераций отдельные
потомки нередко будут содержать митохондрии только одного из родительских типов). В отличие от этого у высших животных яйцеклетка вносит в зиготу больше цитоплазмы чем спермий, а у некоторых животных спермии могут вообще не вносить цитоплазмы. Поэтому можно думать, что у высших животных митохондриальный геном будет передаваться только от одного родителя (а именно по материнской
линии); и действительно, это было подтверждено экспериментами. Оказалось, например, что при скрещивании крыс двух лабораторных линий с митохондриальной ДНК, слегка различающейся по пocледовательности нуклеотидов (типы А и В), получается потомство, содержа-
щее митохондриальную ДНК только материнского типа.
Цитоплазматическая наследственность, в отличие от ядерной, не под-чиняется законам Менделя. Это связано с тем, что у высших животных и расте-ний гаметы от разных полов содержат несопоставимые количества митохон-дрий. Так, в яйцеклетке мыши имеется 90 тыс. митохондрий, а в сперматозоиде — лишь четыре. Очевидно, что в оплодотворенной яйцеклетке митохондрии преимущественно или только от женской особи, т.е. наследование всех мито-хондриальных генов материнское. Генетический анализ цитоплазматической наследственности затруднен из-за ядерно-цитоплазматических взаимодействий. В случае цитоплазматической мужской стерильности мутантный митохон-дриальный геном взаимодействует с определенными генами ядра, рецессивные аллели которых необходимы для развития признака. Доминантные аллели этих генов как в гомо-, так и в гетерозиготном состоянии восстанавливают фертиль-ность растений вне зависимости от состояния митохондриального генома.
Хотелось бы остановиться на механизме материнского наследования генов путем приведения конкретного примера. Для того чтобы окончательно и бесповоротно понять механизм неменделевского (цитоплазматического) наследования митохондриальных генов, рассмотрим, что происходит с такими генами, когда две гаплоидные клетки сливаются, образуя диплоидную зиготу. В случае когда одна дрожжевая клетка несет мутацию, определяющую резистентность митохондриального белкового синтеза к хлорамфениколу, а другая - клетка дикого типа - чувствительна к этому антибиотику: мутантные гены легко выявить, выращивая дрожжи на среде с глицеролом, использовать который способны только клетки с интактными митохондриями; поэтому в присутствии хлорамфеникола на такой среде смогут расти только клетки, несущие мутантный ген. Наша диплоидная зигота вначале будет иметь митохондрии как мутантного, так и дикого типа. От зиготы в результате митоза отпочкуется диплоидная дочерняя клетка, которая будет содержать лишь небольшое число митохондрий. После нескольких митотических циклов в конце концов какая-то из новых клеток получит все митохондрии либо мутантного, либо дикого типа. Поэтому все потомство такой клетки будет иметь генетически идентичные митохондрии. Такой случайный процесс, в результате которого образуется диплоидное потомство содержащее митохондрии только одного типа, называют митотическо
й
се
грегацие
й
.
Когда диплоидная клетка с одним лишь типом митохондрий претерпевает мейоз, все четыре дочерние гаплоидные клетки получают одинаковые митохондриальные гены. Этот тип наследования называют неменде
лев
ским
или цитоплазматическим
в отличие от менделевского наследования ядерных генов. Передача генов по цитоплазматическому типу означает, что изучаемые гены находятся в митохондриях.
Изучение геномов митохондрий, их эволюции, идущей по специфическим законам популяционной генетики, взаимоотношений между ядерными и мито-хондриальными генетическими системами, необходимо для понимания слож-ной иерархической организации эвкариотической клетки и организма в целом.
С определенными мутациями в митохондриальной ДНК или в ядерных генах, контролирующих работу митохондрий, связывают некоторые наслед-ственные болезни и старение человека. Накапливаются данные об участии де-фектов мтДНК в канцерогенезе. Следовательно, митохондрии могут быть ми-шенью химиотерапии рака. Имеются факты о тесном взаимодействии ядерного и митохондриального геномов в развитии ряда патологий человека. Множес-твенные делеции мтДНК обнаружены у больных с тяжелой мышечной слабос-тью, атаксией, глухотой, умственной отсталостью, наследующихся по аутосомно-доминантному типу. Установлен половой диморфизм в клинических проявлениях ишемической болезни сердца, что скорее всего обусловлено мате-ринским эффектом — цитоплазматической наследственностью. Развитие ген-ной терапии внушает надежду на исправление дефектов в геномах митохон-дрий в обозримом будущем.
Как известно, для того чтобы проверить функцию одного из компонентов многокомпонентной системы, необходимой становится ликвидация даного компонента с последующим анализом произошедших изменений. Так как темой даного реферата является указание роли материнского генома для развития потомка, логично было бы узнать о последствиях нарушений в составе митохондриального генома вызванных различными факторами. Инструментом для изучения вышеуказанной роли оказался мутационный процесс, а интересующими нас последствиями его действия стали т.н. митохондриальные болезни.
Митохондриальные болезни представляют собой пример цитоплазмати-ческой наследственности у человека, а точнее «органелльной наследствен-ности». Это уточнение следует сделать,т.к. теперь доказано существование, по крайней мере, у некоторых организмов, цитоплазматических наследственных детерминант, не связанных с клеточными органеллами, - цитогенов(С.Г. Инге-Вечтомов, 1996).
Митохондриальные болезни - гетерогенная группа заболеваний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий и нарушением тканевого дыхания. Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ. Основным биохимическим признаком митохондриальной патологии является развитие лактат-ацидоза, обычно выявляется гиперлактатацидемия в сочетании с гиперпируватацидемией. Число различных вариантов достигло 120 форм. Отмечается стабильное повышение концентрации молочной и пировиноградной кислот в цереброспинальной жидкости.
Митохондриальные болезни (МБ) представляют собой существенную про-блему для современной медицины. По способам наследственной передачи среди МБ выделяют заболевания, наследуемые моногенно по менделевскому типу, при которых в связи с мутацией ядерных генов либо нарушаются структура и функционирование митохондриальных белков, либо изменяется экспрессия митохондриальной ДНК, а также болезни, вызываемые мутациями митохондри-альных генов, которые в основном передаются потомству по материнской линии.
Данные морфологических исследований, свидетельствующие о грубой патологии митохондрий: анормальная пролиферация митохондрий, полимор-физм митохондрий с нарушением формы и размеров, дезорганизация крист, скопления аномальных митохондрий под сарколеммой, паракристаллические включения в митохондрии, наличие межфибриллярных вакуолей
Формы митохондриальных заболеваний
1
. Митохондриальные болезни, вызванные мутациями митохондриальной ДНК
1.1.Болезни, обусловленные делециями митохондриальной ДНК
1.1.1.Синдром Кернса-Сейра
Заболевание проявляется в возрасте 4-18 лет, прогрессирующая наружная офтальмоплегия, пигментный ретинит, атаксия, интенционный тремор, атриовентрикулярная блокада сердца, повышение уровня белка в цереброспи-нальной жидкости более 1 г\л, "рваные" красные волокна в биоптатах скелет-ных мышц
1.1.2.Синдром Пирсона
Дебют заболевания с рождения или в первые месяцы жизни, иногда возможно развитие энцефаломиопатий, атаксии, деменции, прогрессирующей наружной офтальмоплегии, гипопластическая анемия, нарушение экзокринной функции поджелудочной железы, прогрессирующее течение
2
.Болезни, обусловленные точковыми мутациями митохондриальной ДНК
2.1.Наследственная атрофия зрительных нервов Лебера
Материнский тип наследования, острое или подострое снижение остроты зре-ния на один или оба глаза, сочетание с неврологическими и костно-суставными нарушениями, микроангиопатия сетчатки, прогрессирующее течение с возмо-жностью ремиссии или восстановления остроты зрения, дебют заболевания в возрасте 20-30 лет
2.2.Синдром NAPR (невропатия, атаксия, пигментный ретинит)
Материнский тип наследования, сочетание нейропатии, атаксии и пигментного ретинита, задержка психомоторного развития, деменция, наличие "рваных" красных волокон в биоптатах мышечной ткани
2.3.Синдром MERRF (миоклонус-эпилепсия, "рваные" красные волокна)
Материнский тип наследования, дебют заболевания в возрасте 3-65 лет, мио-клоническая эпилепсия, атаксия, деменция в сочетании с нейросенсорной глу-хотой, атрофией зрительных нервов и нарушениями глубокой чувствительно-сти, лактат-ацидоз, при проведении ЭЭГ обследования выявляются генерализо-ванные эпилептические комплексы, "рваные" красные волокна в биоптатах скелетных мышц, прогрессирующее течение
2.4.Синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды)
Материнский тип наследования, дебют заболевания в возрасте до 40 лет, непе-реносимость физических нагрузок, мигренеподобные головные боли с тошно-той и рвотой, инсультоподобные эпизоды, судороги, лактат-ацидоз, "рваные" красные волокна в биоптатах мышц, прогрессирующее течение.
3
.Патология, связанная с дефектами межгеномной коммуникации
3.1.Синдромы множественных делеций митохондриальной ДНК
Блефароптоз, наружная офтальмоплегия, мышечная слабость, нейросенсорная глухота, атрофия зрительных нервов, прогрессирующее течение, "рваные" крас-ные волокна в биоптатах скелетных мышц, снижение активности ферментов дыхательной цепи.
3.2.Синдром делеции митохондриальной ДНК
Аутосомно-рецессивный тип наследования
Клинические формы:
3.2.1.Фатальная инфантильная
а) тяжелая печеночная недостаточность б)гепатопатия в)мышечная гипотония
Дебют в периоде новорожденности
3.2.2.Врожденная миопатия
Выраженная мышечная слабость, генерализованная гипотония, кардиомиопа-тия и судороги, поражение почек, глюкозурия, аминоацидопатия, фосфатурия
3.2.3.Инфантильная миопатия
возникает в первые 2 года жизни, прогрессирующая мышечная слабость, атро-фия проксимальных групп мышц и утрата сухожильных рефлексов, течение быстро прогрессирующее, летальный исход в первые 3 года жизни.
4
.Митохондриальные болезни, обусловленные мутациями ядерной ДНК
4.1.Заболевания, связанные с дефектами дыхательной цепи
4.1.1.Дефицит комлекса 1 (NADH:CoQ-редуктаза)
Начало заболевания до 15 лет, синдром миопатии, задержка психомоторного развития, нарушение сердечно-сосудистой системы, судороги, резистентные к терапии, множественные неврологические нарушения, прогрессирующее тече-ние
4.1.2.Дефицит комплекса 2 (сукцинат-CoQ-редуктаза)
Характеризуется синдромом энцефаломиопатии, прогрессирующие течение, су-дороги, возможно развитие птоза
4.1.3.Дефицит комплекса 3 (CoQ-цитохром С-оксидоредуктаза)
Мультисистемные нарушения, поражение различных органов и систем,с вовле-чением центральной и периферической нервной системы, эндокринной систе-мы, почек, прогрессирующее течение
4.1.4.Дефицит комплекса ( цитохром С-оксидаза)
4.1.4.1.Фатальный инфантильный врожденный лактат-ацидоз
Митохондриальная миопатия с почечной недостаточностью или кардиомиопа-тия, дебют в неонатальном возрасте, выраженные дыхательные нарушения, диффузная мышечная гипотония, течение прогрессирующее, летальный исход на первом году жизни.
4.1.4.2.Доброкачественная инфантильная мышечная слабость
Атрофии, при адекватном и своевременном лечении возможна быстрая стаби-лизация процесса и выздоровление к 1-3 годам жизни
5
.Синдром Менкеса (трихополиодистрофия)
Резкая задержка психомоторного развития, отставание в росте, нарушение рос-та и дистрофические изменения волос,
6
. Митохондриальные энцефаломиопатии
6.1.Синдром Лея
(подострая невротизирующая энцефаломиелопатия)
Проявляется после 6 месяцев жизни, мышечная гипотония, атаксия, нистагм, пирамидные симптомы, офтальмоплегия, атрофия зрительных нервов, часто от-мечается присоединение кардиомиопатии и легкого метаболического ацидоза
6.2.Синдром Альперса
(прогрессирующая склерозирующая полидистрофия)
Дегенерация серого вещества мозга в сочетании с циррозом печени, дефицит комплекса 5 (АТФ-синтетаза), задержка психомоторного развития, атаксия, деменция, мышечная слабость, течение заболевания прогрессирующее, небла-гоприятный прогноз
6.3.Дефицит Коэнзима-Q
Метаболические кризы, мышечная слабость и утомляемость, офтальмоплегия, глухота, снижение зрения, инсультоподобные эпизоды, атаксия, миоклонус-эпилепсия, поражение почек: глюкозурия, аминоацидопатия, фосфатурия, эндо-кринные нарушения, прогрессирующее течение, снижение активности фермен-тов дыхательной цепи
7
.Заболевания, связанные с нарушением метаболизма молочной и пировиноградной кислот
7.1.Дефицит пируваткарбоксилазы Аутосомно-рецессивный тип наследования, дебют заболевания в неоната-льном периоде, симптомокомплекс "вялого ребенка", судороги, резистентные к терапии, высокие концентрации кетоновых тел в крови, гипераммониемия, ги-перлизинемия, снижение активности пируваткарбоксилазы в скелетных мышцах
7.2.Дефицит пируватдегидрогеназы
Проявление в неонатальном периоде, черепно-лицевая дизморфия, судороги, резистентные к терапии, нарушение дыхания и сосания, симптомокомплекс "вя-лого ребенка", дисгинезии мозга, выраженный ацидоз с высоким содержанием лактата и пирувата
7.3.Снижение активности пируватдегидрогеназы
Проявление на первом году жизни, микроцефалия, задержка психомоторного развития, атаксия, мышечная дистония, хореоатетоз, лактат-ацидоз с высоким содержанием пирувата
7.4.Дефицит дигидролипоилтрансацетилазы
Аутосомно-рецессивный тип наследования, дебют заболевания в неонатальном периоде, микроцефалия, задержка психомоторного развития, мышечная гипотония с последующим повышением мышечного тонуса, атрофия дисков зрительных нервов, лактат-ацидоз, снижение активности дигидролипоилтранс-ацетилазы
7.5.Дефицит дигидролипоилдегидрогеназы
Аутосомно-рецессивный тип наследования, дебют заболевания на первом году жизни, симптомокомплекс "вялого ребенка", дисметаболические кризы со рво-той и диареей, задержка психомоторного развития, атрофия дисков зрительных нервов, лактат-ацидоз, повышение содержания в сыворотке крови аланина, α-кетоглутарата, α-кетокислот с разветвленной цепью, снижение активности ди-гидролипоилдегидрогеназы
8
.Заболевания, обусловленные дефектами бета-окисления жирных кислот
8.1.Недостаточность Ацетил-CoA-дегидрогеназы с длинной углеродной цепью
Аутосомно-рецессивный тип наследования, дебют заболевания в первые месяцы жизни, метаболические кризы со рвотой и диареей, симптомокомплекс "вялого ребенка", гипогликемия, дикарбоксиловая ацидурия, снижение актив-ности ацетил-CoA-дегидрогеназы жирных кислот с длинной углеродной цепью
8.2.Недостаточность Ацетил-CoA-дегидрогеназы со средней углеродной цепью
Аутосомно-рецессивный тип наследования, дебют заболевания в неонатальном периоде или первые месяцы жизни, метаболические кризы со рвотой и диареей,
мышечная слабость и гипотония, часто развивается синдром внезапной смерти, гипогликемия, дикарбоксиловая ацидурия, снижение активности ацетил-CoA-дегидрогеназы жирных кислот со средней углеродной цепью
8.3. Недостаточность Ацетил-CoA-дегидрогеназы жирных кислот с короткой углеродной цепью
Аутосомно-рецессивный тип наследования, различный возраст дебюта заболевания, снижение толерантности к физическим нагрузкам, метаболичес-кие кризы со рвотой и диареей, мышечная слабость и гипотония, увеличение экскреции с мочой метилсукциновой кислоты, ацетил-CoA-дегидрогеназы жирных кислот с короткой углеродной цепью
8.4.Множественная недостаточность Ацетил-CoA-дегидрогеназ жирных кислот
Неонатальная форма
: черепно-лицевая дизморфия, дисгинезии мозга, тяжелая гипогликемия и ацидоз, злокачественное течение, снижение активности всех ацетил-СоА-дегидрогеназ жирных кислот,
Инфантильная форма:
симптомокосплекс "вялого ребенка", кардиомиопатия, метаболические кризы, гипогликемия и ацидоз
8.5.Снижение активности всех ацетил-СоА-дегидрогеназ жирных кислот
Форма с поздним дебютом:
периодические эпизоды мышечной слабости, мета-болические кризы, гипогликемия и ацидоз менее выражены, интеллект сохра-нен,
9
.Ферментопатии цикла Кребса
9.1.Дефицит фумаразы
Аутосомно-рецессивный тип наследования, дебют заболевания в неонатальном периоде или периоде новорожденности, микроцефалия, генерализованная мы-шечная слабость и гипотония, эпизоды летаргии, быстро прогрессирующая эн-цефалопатия, неблагоприятный прогноз
9.2.Дефицит сукцинатдегидрогеназы
Редкое заболевание, характеризующееся прогрессирующей энцефаломиопатией
9.3.Дефицит альфа-кетоглутаратдегидрогеназы
Аутосомно-рецессивный тип наследования, неонатальный дебют заболевания, микроцефалия, симптомокомплекс "вялого ребенка", эпизоды летаргии, лактат-ацидоз, быстро прогрессирующее течение, снижение содержания ферментов цикла Кребса в тканях
9.4.Синдромы дефицита карнитина и ферментов его метаболизма
Дефицит карнитин-пальмитоилтрансферразы-1, аутосомно-рецессивный тип наследования, ранний дебют заболевания, эпизоды не кетонемической гипогли-кемической комы, гепатомегалия, гипертриглицеридемия и умеренная гиперам-мониемия, снижение активности карнитин-пальмитоилтрансферразы-1 в фибробластах и клетках печени
9.5.Дефицит карнитин-ацилкарнитин-транслоказы
Ранний дебют заболевания, сердечно-сосудистые и дыхательные нарушения, симптомокомплекс "вялого ребенка", эпизоды летаргии и комы, повышение концентрации эфиров карнитина и длинной углеродной цепью на фоне сниже-ния свободного карнитина в сыворотке крови, снижение активности карнитин-ацилкарнитин-транслоказы
9.6.Дефицит карнитин-пальмитоилтрансферразы-2
Аутосомно-рецессивный тип наследования, мышечная слабость, миалгии, миоглобинурия, снижение активности карнитин-пальмитоилтрансферразы-2 в скелетных мышцах
9.7.Почечный дефект транспортной системы карнитина
Аутосомно-рецессивный тип наследования, миопатический симптомокомплекс, эпизоды вялости и летаргии, кардиомиопатия, эпизоды гипогликемии, снижение уровня карнитина в сыворотке крови и увеличение его экскреции с мочой.
Проанализировав такой ‘страшный’ список патологий, связанных с теми или другими изменениями функционирования митохондриального(и не только) генома возникают определенные вопросы. Что же собой представляют продукты митохондриальных генов и в каких именно супермега-жизненноважных клеточных процессах они принимают участие?
Как оказалось, некоторые из вышеперечисленных патологий могут возни-кать при нарушениях синтеза 7 субъединиц НАДН-дегидрогеназного комплек-са, 2 субъединиц АТФ-синтетазы, 3 субъединиц цитохром-с-оксидазы и 1 субъединицы убихинол-цитохром-с-редуктазы(цитохром b), которые и являют-ся генными продуктами митохондрий. Исходя из этого можно сделать вывод о существовании ключевой роли данных белков в процессах клеточного дыхания, окисления жирных кислот и синтеза АТФ, переноса электронов в электронтран-спортной системе внутренней мт мембраны, функционирования антиоксидант-ной системы и т.д.
Судя по последним данным о механизмах апоптоза, многие ученые пришли к выводу о наличии центра контроля апоптоза именно в лице митохондрий...
Роль митохондриальных белков также была показана при применении антибиотиков, блокирующих мт синтез. Если клетки человека в культуре ткани обработать антибиотиком, например тетрациклином или хлорамфениколом, то после одного-двух делений их рост прекратится. Это связано с ингибированием митохондриального белкового синтеза, приводящим к появлению дефектных митохондрий и как следствие к недостаточному образованию АТР. Почему же тогда антибиотики можно использовать при лечении бактериальных инфекций? Есть несколько ответов на этот вопрос:
1. Некоторые антибиотики (такие, как эритромицин) не проходят через внутрен-нюю мембрану митохондрий млекопитающих.
2. Большинство клеток нашего тела не делятся или делятся очень медленно, поэтому столь же медленно происходит и замена существующих митохондрий новыми (во многих тканях половина митохондрий заменяется примерно за пять дней или еще дольше). Таким образом, количество нормальных митохондрий снизится до критического уровня только в том случае, если блокада митохондриального белкового синтеза будет поддерживаться на протяжении многих дней.
3. Определенные условия внутри ткани препятствуют проникновению некоторых препаратов в митохондрии наиболее чувствительных клеток. Например, высокая концентрация Са2+
в костном мозге приводит к образованию Са2+
-тетрациклинового комплекса, который не может проникнуть в быстро делящиеся (и потому наиболее уязвимые) предшественники клеток крови.
Эти факторы дают возможность использовать некоторые препараты, ингиби-рующие митохондриальный синтез белка, в качестве антибиотиков при лечении высших животных. Только два таких препарата оказывают побочное действие: длительное лечение большими дозами хлорамфеникола может привести к нарушению кроветворной функции костного мозга (подавить образование эритроцитов и лейкоцитов), а длительное применение тетрациклина - к поврежде-нию кишечного эпителия. Но в обоих случаях еще не вполне ясно, вызываются ли эти побочные эффекты блокадой биогенеза митохондрий или какими-то иными причинами.
Вывод
Структурно-функциональные особенности мт генома состоят в следу-ющем. Во-первых, установлено, что мтДНК передается от матери всем ее
потомкам и от ее дочерей всем последующим поколениям, но сыновья не передают свою ДНК (материнское наследование). Материнский характер
наследования мтДНК, вероятно, связан с двумя обстоятельствами: либо доля отцовских мтДНК так мала (по отцовской линии может передаваться не
более одной молекулы ДНК на 25 тыс. материнских мтДНК), что они не могут быть выявлены существующими методами, либо после оплодотворения блоки-руется репликация отцовских митохондрий. Во-вторых, отсутствие комбинати-вной изменчивости — мтДНК принадлежит только одному из родителей, сле-довательно рекомбинационные события, характерные для ядерной ДНК в мейо-зе, отсутствуют, а нуклеотидная последовательность меняется из поколения в поколение только за счет мутаций. В-третьих, мтДНК не имеет интронов
(большая вероятность, что случайная мутация поразит кодирующий район ДНК), защитных гистонов и эффективной ДНК-репарационной системы —все это определяет в 10 раз более высокую скорость мутирования, чем в ядерной ДНК. В-четвертых, внутри одной клетки могут сосуществовать одновременно нормальные и мутантные мтДНК —явление гетероплазмии (присутствие толь-ко нормальных или только мутантных мтДНК называется гомоплазмией). Наконец, в мтДНК транскрибируются и транслируются обе цепи, а по ряду ха-рактеристик генетический код мтДНК отличается от универсального (UGA кодирует триптофан, AUA кодирует метионин, AGA и AGG являются стоп-
кодонами).
Эти свойства и вышеуказанные функции мт-генома сделали иссле-дование изменчивости нуклеотидной последовательности мтДНК неоценимым инструментом для врачей, судебных медиков, биологов-эволюционистов,
представителей исторической науки в решении своих специфических задач.
Начиная с 1988 г., когда было открыто, что мутации генов мтДНК лежат в основе митохондриальных миопатий (J.Y. Holt et al., 1988) и наследственной оптической нейропатии Лебера (D.C. Wallace, 1988), дальнейшее систематичес-кое выявление мутаций мт-генома человека привело к формированию концеп-ции митохондриальных болезней (МБ). В настоящее время патологические му-тации мтДНК открыты в каждом типе митохондриальных генов.
Список литературы
1. Скулачев В.П. Эволюция, митохондрии и кислород, Сорос. образоват. журн.
2. Ленинджер А. Основы биохимии: В трех томах, М.: Мир, 1985-1986.
3. Nicholes D.G. Bioenergetics, An Introd. to the Chemiosm. Th., Acad. Press, 1982.
4. Stryer L. Biochemistry, 2nd ed. San Fransisco, Freeman, 1981.
5. Скулачев В.П. Энергетика биологических мембран. М., 1989.
6. Бакеева Л.Е., Ченцов Ю.С. Митохондриальный ретикулум: Строение и некоторые функции // Итоги науки. Общие проблемы биологии. 1989
7. Ченцов Ю.С. Общая цитология. М.: Изд-во МГУ, 1995
8. Пузырев В.П., Голубенко М.В., Фрейдин М.Б.
Сфера компетенции митохон-дриального генома // Вестн. РАМН, 2001. ‹ 10. С. 31—43.
9. Holt I.J, Harding A.E., Morgan-Hughes I.A. Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature, 1988, 331:717-719.
10.
Баранов В.С. и др.
Геном человека и гены предрасположенности. СПб., 2000
11. Минченко А.Г., Дударева Н.А.
Митохондриальный геном. Новосибирск, 1990.
12. Гвоздев В.А.
// Сорос. образоват. журн. 1999. №10. С.11—17.
13. Маргелис Л.
Роль симбиоза в эволюции клетки. М., 1983.
14. Скулачев В.П.
// Сорос. образоват. журн. 1998. №8. С.2—7.
15. Игамбердиев А.У.
// Сорос. образоват. журн. 2000. №1. С.32—36.
Киевский Национальный Университет им. Тараса Шевченка
Биологический факультет
Реферат
на тему:
“Роль материнского генома в развитии потомка”
с
туд
е
нта
IV
курса
кафедры биохимии
Фролова Артема
Киев
2004
План
:
Вступление...............................................................................1
Симбиотическая теория происхождения митохондрий......2
Роль клеточного ядра в биогенезе митохондрий...................................5
Транспортные системы митохондрий.....................................................7
Размеры и форма митохондриальных геномов..................10
Функционирование митохондриального генома...............14
Значение наличия собственной генетической системы для митохондрий..............................................................................19
Цитоплазматическая наследственность..............................20
|