| Министерство образования и науки Российской Федерации
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования «Северо-Кавказский государственный технический университет»
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
к выполнению лабораторных работ по дисциплине «Аналитическая химия и физико-химические методы анализа» для студентов специальностей 240901 «Биотехнология»;
240902 «Пищевая биотехнология»;
260202 «Технология хлеба, кондитерских и макаронных изделий»;
260301 «Технология мяса и мясных продуктов»;
260303 «Технология молока и молочных продуктов»;
240403 «Химическая технология природных энергоносителей и углеродных материалов»;
240306 «Химическая технология монокристаллов, материалов и изделий электронной техники»;
240100 «Химическая технология и биотехнология – бакалавриат»
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Ставрополь
2009
Методические указания «Физико-химические методы анализа» по дисциплине «Аналитическая химия и физико-химические методы анализа» для подготовки и выполнения лабораторных работ содержат теоретический материал, подробную методику выполнения работы, форму отчета, а также контрольные вопросы, перечень литературы и подразумевают индивидуальную работу студентов по подготовке, выполнению и защите лабораторных работ.
Методические указания составлены в соответствии с учебным планом и программой дисциплины.
Составители: О. А. Слепышева, канд. хим. наук, доцент,
М. В. Грицаева, ассистент
Рецензент: С. Э. Хорошилова, канд. хим. наук, доцент
СОДЕРЖАНИЕ
Хроматографический метод анализ
Лабораторная работа 18.
Определение состава смеси органических спиртов 4
Оптические методы анализа
Лабораторная работа 19.
Определение содержания ионов металлов в растворе фотоколориметрическим методом 20
Лабораторная работа 20.
Определение содержания лактозы
(сахарозы) методом рефрактометрии 28
Электрохимические методы анализа
Лабораторная работа 21.
Определение содержания карбоната и гидрокарбоната в смеси методом прямого потенциометрического титрования 37
Список рекомендуемой литературы
48 ХРОМАТОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗ
ЛАБОРАТОРНАЯ РАБОТА 18
ОПРЕДЕЛЕНИЕ СОСТАВА СМЕСИ ОРГАНИЧЕСКИХ СПИРТОВ
1 Цель и содержание
Получить представление о методе газожидкостной хроматографии (ГЖХ). Научиться определять качественный и количественный состав смеси органических веществ по хроматограмме.
2 Теоретическое обоснование
Хроматографический метод
– это метод разделения смесей газов, паров, жидкостей и растворенных веществ с помощью динамической сорбции
. Под сорбцией понимается поглощение газов, паров или растворенных веществ твердыми или жидкими поглотителями. Сорбция – общее понятие, включающее в себя адсорбцию (поглощение на поверхности фазы) и абсорбцию (поглощение в объеме фазы).
Различают физическую адсорбцию
и химическую адсорбцию
, или хемосорбцию
. В первом случае адсорбционные силы имеют ту же природу, что и межмолекулярные или ван-дер-ваальсовы силы. Это дальнодействующее, но слабое взаимодействие, и количество энергии, выделяющееся при физической адсорбции, находится в пределах 20 кДж
/ моль
. Физическая адсорбция всегда обратима.
При химической адсорбции (хемосорбции) молекулы удерживаются на поверхности в результате образования химической, и обычно ковалентной, связи. Энергия связи в данном случае значительно больше, чем при физической адсорбции, и обычные значения находятся в области 200 кДж
/ моль
. Хемосорбция обычно необратима. Следует отметить, что провести резкую границу между обоими видами адсорбции невозможно.
Поглощающие вещества называются сорбентами
, а поглощаемые – сорбатами
. Процесс, обратный сорбции, называют десорбцией.
В зависимости от типа физико-химического взаимодействия между сорбентом и находящимся в растворе веществом различают 3 типа хроматографии: адсорбционную, ионообменную и распределительную.
Адсорбционная хроматография
основана на количественном различии в адсорбционных свойствах компонентов разделяемой смеси, причем в качестве адсорбента используется твердый материал (твердая фаза), на поверхности которого идет процесс сорбции. Зная, что адсорбционное сродство полярных веществ к полярным адсорбентам гораздо выше, чем неполярных, можно легко понять, почему гидрофильный (т. е. легко смачивающийся водой) полярный силикагель используют для осушения масел, бензина, воздуха, а неполярный гидрофобный (т. е. легко смачивающийся неполярным углеводородом) активированный уголь используют для очистки воды от примесей нефтепродуктов.
Ионообменная хроматография
основана на обратимом стехиометрическом обмене ионов, содержащихся в хроматографируемом растворе, на подвижные ионы, входящие в состав ионитов (ионообменников).
Распределительная хроматография
основана на количественном различии в коэффициентах распределения компонентов разделяемой смеси между неподвижной и подвижной несмешивающимися фазами.
В зависимости от природы подвижной фазы хроматографические методы можно разделить на два основных типа: газовую хроматографию и жидкостную хроматографию.
Газовая хроматография
– это метод разделения летучих веществ пропусканием газового потока через неподвижную фазу. Известны два основных вида хроматографии: ГЖХ (газо-жидкостная хроматография) и ГАХ (газо-адсорбционная хроматография).
Газо-адсорбционная хроматография
– это вариант хроматографии, в котором разделение производится с помощью подвижной газовой фазы, проходящей вместе с анализируемой смесью над твердым сорбентом. В качестве сорбента используют силикагель, молекулярные сита, пористые полимеры и т. д. Более подробно рассмотрим метод газо-жидкостной хроматографии
, в котором неподвижной фазой служит нелетучая жидкость, нанесенная в виде пленки на твердый сорбент. Основу метода ГЖХ составляет процесс распределения веществ между двумя фазами: неподвижной (нелетучей жидкостью) и подвижной (газом-носителем), осуществляемый в аналитическом приборе, называемом газовым хроматографом (рис. 18.1).

Рисунок 18.1 – Принципиальная схема газового хроматографа:
1
– блок подготовки газов, в котором осуществляется очистка газов от микропримесей воды, кислорода и органических веществ, а также регулируются
давления и расходы; 2 –
испаритель, т. е. устройство ввода пробы и перевода ее в
парообразное состояние; 3
– термостат колонок; 4
– детектор (в данном случае ионизационно-пламенный); 5
– усилитель; 6 –
самописец
В комплект современного газо-жидкостного хроматографа всегда входит компьютер, принтер и т. д. Унифицированный пакет прикладных программ позволяет полностью автоматизировать весь аналитический процесс от пробоподготовки до распечатки результатов с учетом конкретных задач и требований.
Анализируемая проба (смесь летучих веществ) с помощью микрошприца вводится в испаритель, в котором обычно устанавливается температура на 20 − 30o
С
выше температуры кипения самого высококипящего компонента. В испарителе проба быстро переводится в газообразное состояние и в потоке газа-носителя поступает в колонку. Проходя вместе с газом-носителем через колонку, газообразные компоненты пробы распределяются между подвижной фазой (газ-носитель) и неподвижной жидкой фазой, покрывающей тонким слоем твердый носитель в колонке
(рис. 18.2).
В качестве неподвижной жидкой фазы обычно используют силиконовые масла, каучуки, высокомолекулярные полиэфиры и т. д. Температура хроматографической колонки поддерживается приблизительно наполовину ниже температуры кипения наиболее высококипящего вещества в смеси для обеспечения актов сорбции–десорбции. Эффективность хроматографического разделения, прежде всего, зависит от скорости миграции (т. е. перемещения) молекул исследуемого вещества через колонку.
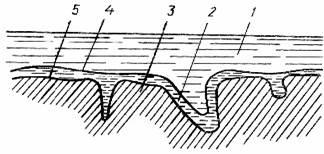
Рисунок 18.2 – Схема строения слоя сорбента в газо-жидкостной хроматографии:
1 – подвижная фаза (газ); 2 – неподвижная жидкая фаза; 3 – твердый носитель; 4 – поверхность раздела газ-неподвижная; 5 –жидкая фаза
Скорость миграции зависит от распределения компонента между неподвижной и подвижной фазами, т. е. от константы распределения. Большое внимание уделяется и размыванию хроматографических зон, вызываемому неравновесностью распределения вещества между неподвижной и подвижной фазами, диффузией в газовой фазе и т. д. Таким образом, различные по природе компоненты, составляющие пробу, движутся с различными скоростями, разделяются на отдельные зоны и элюируются (т. е. проходят вместе с газом-носителем) один за другим в виде более или менее широких зон через детектор. Как видно из рисунка 18.3, подвижная фаза (Е) с постоянной скоростью протекает через хроматографическую колонку. Определенное количество парообразной пробы вводится в подвижную фазу перед входом в колонку в виде небольшой «пробки». В колонке отдельные компоненты неодинаково долго удерживаются неподвижной фазой. Благодаря этому они продвигаются по колонке медленнее, чем подвижная фаза, и с различными скоростями. Поэтому первоначальная «пробка» постепенно расщепляется на несколько зон (рис. 18.3).

Рисунок 18.3 – Образование зон в процессе хроматографирования смеси
Детектор реагирует на присутствие анализируемого соединения в потоке газа-носителя и посылает электрический сигнал через усилитель к самописцу. Таким образом, каждое соединение дает пик, записываемый на ленте самописца. Запись результатов хроматографического анализа самописцем на диаграммной ленте в виде пиков (пропорциональных концентрации веществ в газе-носителе) называется дифференциальной хроматограммой.
Параметры хроматограммы
В хроматограмме на оси абсцисс регистрируется время выхода из колонки каждого компонента пробы, а на оси ординат — сигналы детектора, пропорциональные содержанию компонента в пробе (рис. 4). В методе хроматографии основным количественным соотношением, характеризующим скорость миграции молекул исследуемого вещества через колонку, служит время удерживания — t
R
.
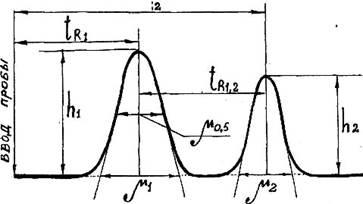
Рисунок 18.4 – Параметры дифференциальной хроматограммы:
t
R
1
и t
R
2
– время удерживания компонентов 1 и 2; h
1
и h
2
– высоты пиков;
m
0,5
– ширина пика на середине высоты; m
– ширина пика у основания;
∆t
R
1, 2
– интервал между максимумами двух соседних пиков
Время удерживания
– это характеристика сорбционной способности неподвижной фазы по отношению к разделяемым веществам при данных условиях хроматографирования. Практически время удерживания определяется расстоянием на хроматограмме от момента ввода пробы до момента появления на хроматограмме компонента в максимальной концентрации. По мере перемещения анализируемых веществ по колонке происходит их постепенное разбавление газомносителем и формирование непрерывно расширяющихся зон. Расширение зон препятствует разделению анализируемых веществ, а разбавление компонентов часто затрудняет обнаружение пиков.
В хроматографическом анализе принято связывать эффективность хроматографической колонки с расширением зоны анализируемого вещества, а основным количественным выражением, характеризующим эффективность хроматографического разделения, является высота (Н) эквивалентной теоретической тарелки (ВЭТТ).
Значение ВЭТТ рассчитывают по формуле:
H
=  L
, (18.1) L
, (18.1)
N
где L
−длина хроматографической колонки, см
; N
−число теоретических тарелок.
Согласно теории ВЭТТ хроматографическая колонка предполагалась состоящей из воображаемых тарелок, на которых происходило достижение полного равновесия анализируемого вещества между двумя фазами. Тогда высокая эффективность колонки проявляется в большом числе N
таких теоретических тарелок.
Для определения N
обычно используется следующее уравнение:
N
= 5,54
 m
t
0R
,5
2
. (18.2) m
t
0R
,5
2
. (18.2)
Следует отметить, что чем меньше высота H
, тем эффективнее работает колонка, т. е. тем более узкими становятся пики на хроматограмме.
Количественный анализ
Для количественной оценки качества хроматографического разделения используют критерий разделения
R
, который учитывает воздействие на полноту разделения компонентов таких факторов, как эффективность колонки и селективность насадки. Параметр R
рассчитывают по формуле (18.3). Он может принимать значения от 0 до ∞. При значении R
=1 происходит полное разделение компонентов.
2∆t
R
1, 2
R
=  . (18.3) m
1 + m
2 . (18.3) m
1 + m
2
Количественный газохроматографический анализ основан на допущении, что площади пиков (или высоты), соответствующие индивидуальным соединениям на хроматограмме, пропорциональны их количеству или концентрации. Площади пиков S
на хроматограмме обычно измеряют методом триангуляции (приближения к площади треугольника) по формуле: S
= h
⋅ m
0,5
, где h
– высота пика, мм
; а m
0,5
– ширина пика на середине его высоты, мм
.
В практике газо-жидкостной хроматографии применяются следующие методы количественного анализа:
- метод абсолютной калибровки;
- метод внутренней нормализации; - метод внутреннего стандарта.
Метод
абсолютной калибровки
заключается в построении графической зависимости одного из количественных параметров хроматографического пика (высоты или площади) от содержания вещества в пробе. Важнейшими требованиями к эксперименту при выполнении количественного анализа по методу абсолютной калибровки являются:
1) точность и воспроизводимость дозирования пробы;
2) строгое соблюдение постоянства условий хроматографирования при калибровке прибора и при определении содержания интересующего вещества в пробе неизвестного состава.
Следует отметить, что воспроизводимость и, соответственно, точность результатов при дозировании жидких проб микрошприцем определяется во многом субъективными факторами, присущими оператору.
Метод внутренней нормализации
предусматривает отнесение измеренной величины количественного параметра хроматографического пика (площади S
i
, или высоты H
i
) к величине суммарного сигнала детектора на все компоненты, присутствующие в анализируемом образце. При этом концентрацию компонентов в анализируемой смеси C
i
(%) находят по формуле:
C
i
=  n
f
i
⋅
C
i
, (18.4) n
f
i
⋅
C
i
, (18.4)
∑ f
i
⋅C
i
i
=1
где S
i
−площадь хроматографического пика; f
i
−калибровочный множитель, учитывающий неодинаковую чувствительность детектора к анализируемым веществам.
Наиболее распространенный способ экспериментального определения f
i
, для какого-либо вещества относительно стандартного соединения заключается в хроматографировании искусственно составленных смесей необходимых компонентов с выбранным стандартным веществом и последующем расчете по формуле:
f
i
=  C
i
⋅
S
ст
, (18.5) C
i
⋅
S
ст
, (18.5)
С
ст
⋅ S
i
где C
i
− концентрация ( масс
.%) i
-го компонента; S
i
− площадь хроматографического пика i
-го компонента; C
ст
− концентрация ( масс
.%) стандартного соединения; S
ст
− площадь хроматографического пика стандартного соединения.
Преимущества метода внутренней нормализации по сравнению с методом абсолютной калибровки заключаются в устранении необходимости точной дозировки образца и соблюдения тождественности условий анализа при повторных определениях. Существенным недостатком этого метода является то, что определение i
-го компонента в сложной смеси правомерно лишь при условии, что природа всех интересующих соединений в смеси известна и все они проявляются на хроматограмме.
Метод внутреннего стандарта
предусматривает прибавление к известному количеству анализируемого вещества также известного количества не содержащегося в нем эталонного соединения («внутреннего стандарта») с последующим хроматографированием смеси. Процентную (масс. или об.) концентрацию компонентов в анализируемом образце С
i
находят по формуле:
C
i
=  S
i
⋅
f
i
⋅
m
ст
, (18.6) S
i
⋅
f
i
⋅
m
ст
, (18.6)
S
ст
⋅ m
см
где S
i
и S
ст
− площади хроматографических пиков определяемого и стандартного веществ; f
i
− калибровочный множитель для определяемого соединения относительно стандартного вещества; m
ст
и m
см
− массы внутреннего стандарта и анализируемой смеси.
Достоинство метода состоит в том, что отпадает необходимость дозирования строго заданных количеств пробы и соблюдения постоянства всех параметров хроматографирования. Ограничения метода заключаются в трудности выбора внутреннего стандарта и в необходимости специальной подготовки пробы для анализа.
3 Приборы и реактивы
Хроматограф: Цвет-100 с детектором ионизационно-пламенным; хроматографическая колонка: стеклянная длиной L
= 3 м
, диаметром
d
= 3 мм
; твердый носитель: хроматон N-AW-DMCS зернения 0,125 − 0,160 мм
; неподвижная жидкая фаза: полиэтиленгликоль− 400 (15% от массы
твердого носителя); микрошприц вместимостью 10 мкл
; анализируемый раствор – смесь жидких спиртов.
4 Указания по технике безопасности
1. При работе в химической лаборатории необходимо соблюдать правила безопасной работы, порядок и чистоту на рабочем месте. При любых выявленных нарушениях техники безопасной работы или ее необеспечения на рабочем месте немедленно поставить в известность преподавателя или лаборанта.
2. Верхняя одежда и личные вещи во время работы в аудитории должны находиться в специально отведенном для этих целей месте.
3. Запрещено выполнять работы в лаборатории, если в аудитории менее 3-х человек или в отсутствие преподавателя (лаборанта). При несчастном случае необходимо немедленно поставить в известность преподавателя и оказать первую помощь пострадавшему.
4. К работе допускаются студенты, имеющие конспект выполняемой работы и знающие правила и приемы безопасного ее выполнения.
5. Работать нужно в халате, избегая попадания в глаза, на руки или одежду химических реактивов. По окончании работы тщательно убрать рабочее место, вымыть посуду и руки.
6. Категорически запрещено принимать как в аудитории, так и во время выполнения работы какую-либо пищу.
7. Все работы с концентрированными кислотами, щелочами, летучими веществами проводить только в вытяжном шкафу. Не сливать в раковины концентрированные растворы, а собирать их в специальную посуду.
8. При разливе кислот и щелочей их надо немедленно нейтрализовать, а место – тщательно промыть.
9. При попадании на стол растворов реактивов, солей или воды, его необходимо протереть сначала влажной тряпкой, а потом сухой. Затем тщательно вымыть руки и убедиться, что растворы не попали на одежду.
10. При работе со стеклянной посудой (колбы, пипетки, мензурки и т. д.) во избежание порезов обращать внимание на их целость. Мытье пробирок и колб выполнять с использованием ершиков.
11. Стеклянные пробки для склянок ставить только на торец, чтобы исключить попадания реактивов на стол. При отборе пробы не допускать попадания капель на внешние стенки склянок, колб, пробирок и другой посуды.
12. Работать надо только на исправном электрооборудовании и газовых горелках, строго соблюдая правила их эксплуатации. В случае обнаружения повреждений немедленно прекратить работу, поставить в известность преподавателя и отключить электроприборы или подачу газа.
13. При нагреве жидкостей на газовой горелке использовать треногу с сеткой, применять для этих целей только специальную термостойкую посуду (маркировка в виде матового квадрата или круга и буквы ТС
). Не допускать бурного кипения и разбрызгивания растворов.
14. При нагреве пробирок использовать специальный держатель, а заполнять их не более, чем на 1/3 объема. В ходе нагрева держать отверстие пробирки так, чтобы пары не попадали на соседей или студентов напротив. В случае термического ожога – удалить с поверхности кожи химические реактивы и оказать первую медицинскую помощь.
15. При возникновении пожара немедленно прекратить работу и поставить в известность преподавателя. Затем выключить все газовые приборы и электрооборудование, вынести, по-возможности, из аудитории горючие и легко воспламеняющиеся жидкости. При тушении использовать песок, кошму, порошковый огнетушитель. Летучие жидкости с малой плотностью нельзя тушить водой.
5 Методика и порядок выполнения работы
1. Подготовка к анализу
1.1. Ознакомиться с описанием, устройством и порядком работы газожидкостного хроматографа, методикой обработки хроматограммы (см. теоретическую часть).
1.2. Получить у преподавателя вариант контрольной хроматограммы смеси спиртов и перенести ее в конспект работы.
2. Качественный анализ смеси спиртов
2.1. Выполнить обработку хроматограммы, как это показано на рис. 18.4 для каждого компонента.
2.2. Определить по хроматограмме время выхода t
R
каждого компонента смеси (в мм) и рассчитать время удерживания (c
) по формуле:
∆
l
t
R
= ⋅60, V
где ∆l
−расстояние с момента ввода пробы до максимума соответствующего пика, мм
; V
−скорость движения ленты самописца. В расчетах принять значение V
=15 мм
/ мин
.
2.3. Выполнить качественную идентификацию спиртов полученной смеси, используя справочные данные таблицы 18.1.
Таблица 18.1 – Время удерживания спиртов

3. Количественный анализ смеси спиртов
3.1 Определить высоту h
и ширину пика m
в мм для каждого компонента.
3.2. Измерить и записать значение m
0,5
на половине высоты хроматографического пика в мм
.
3.3. Определить эффективность работы хроматографической системы для двух, заданных преподавателем, компонентов, рассчитав для них значение ВЭТТ в мм
и число N
теоретических тарелок по формулам:
для ВЭТТ : H
= N
L
и N
= 5,54 m
t
0R
,5 2
.
В расчете использовать значение L
= 300 см
и t
R
.в мм
.
3.4. На основании расчетов N
и ВЭТТ сделать вывод об эффективности работы колонки в целом и по каждому компоненту.
3.5. Для этих же компонентов определить критерий разделения R
по формуле:
2∆t
R
1, 2
R
= . m
1 + m
2
3.6. Исходя из значения R
, сделать вывод о качестве разделения компонентов (ответ – обосновать).
3.7. Методом триангуляции (площади треугольника) по значениям h
и µ0,5
рассчитать площади пиков для всех компонентов смеси: S
= h
⋅ m
0,5
.
3.6. Определить объемное процентное содержание по каждому компоненту X
i
(об
.%):
X
i
(%) =  S
i
. S
i
.
S
1 + S
2 + S
3 + S
4 + S
5
Запись данных опыта.
Все расчеты и выводы привести в отчете по работе, а результаты измерений и вычислений занести в таблицу 18.2.
Таблица 18.2 – Расчет состава смеси органических спиртов
| №
пика
|
определяемый
компонент
|
t
R
|
h
,
мм
|
m
0,5 , мм
|
S
, мм
2
|
X
, %
|
m
,
мм
|
| мм
|
с
|
| 1
|
| 2
|
| 3
|
| 4
|
| 5
|
| |
6 Содержание отчета и его форма
Каждый студент должен иметь лабораторный журнал, который является документом, отражающим всю его работу.
Все наблюдения и выводы по экспериментальной работе, проделанной в лаборатории, студент заносит в лабораторный журнал непосредственно после ее выполнения в виде отчета.
Рекомендуется следующая схема записи:
1. Дата.
2. Номер и название лабораторной работы.
3. Цель работы.
4. Название и описание хода опытов.
5. Наблюдения.
6. Уравнения химических реакций.
7. Выводы и расчеты (формулы, таблицы, графики).
7 Вопросы для защиты работы
1. В чем сущность методов хроматографии?
2. Назовите основные виды адсорбции. В чем заключается их существенное отличие?
3. В чем отличие процессов адсорбции и абсорбции?
4. Перечислите возможности применения метода ГЖХ.
5. Дайте классификацию хроматографических методов по аппаратурному оформлению, агрегатному состоянию фаз и методике хроматографирования.
6. Приведите принципиальную схему ГЖХ-хроматографа и поясните принцип его работы.
7. Параметры, характеризующие хроматограмму и их суть.
8. Что характеризует критерий разделения и ВЭТТ; число N
?
9. Как охарактеризовать эффективность работы хроматографической колонки?
10. Какой параметр хроматограммы позволяет проводить качественный анализ смеси и как его определить?
11. Перечислите основные методы количественной оценки хроматограмм; приведите расчетные формулы.
12. Назовите достоинства и недостатки основных методов количественного анализа в хроматографии.
8 Литература
[1, 2, 3, 4, 5, 6, 9, 10, 11, 12, 16]
ОПТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
ЛАБОРАТОРНАЯ РАБОТА 19
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ИОНОВ МЕТАЛЛОВ В РАСТВОРЕ ФОТОКОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
1 Цель и содержание
Изучить основы метода ФЭК, принцип работы и методику измерений оптической плотности растворов с помощью фотоэлектроколориметра. Освоить методику построения градуировочного графика и определения по нему содержания вещества в пробе.
2 Теоретическое обоснование
Фотоколориметрия относится к оптическим методам анализа. В основе расчетов лежит закон Бугера-Ламберта-Бера:
А
= −e
Cl
,
где А
−оптическая плотность, e
−молярный коэффициент светопоглощения при данной длине волны l
монохроматического излучения, С
−молярная концентрация раствора, моль
/ л
, l
−толщина поглощающего слоя в сантиметрах.
Растворы для фотометрирования должны отвечать следующим условиям: иметь интенсивную окраску, быть разбавленными и прозрачными, чтобы исключить рассеяние света; устойчивыми (не расслаиваться), в них не должно протекать фотохимических и других реакций, влияющих на ход анализа.
Аналитическую длину волны (l
) выбирают путем фотометрирования одного и того же раствора при разных светофильтрах. Оптимальное значение λ соответствует наибольшему светопоглощению анализируемого раствора, при этом цвет светофильтра должен дополнять окраску анализируемого раствора до белой (принцип дополнительности). При правильном подборе условий анализа градуировочный график представляет собой прямую линию в координатах A
− C
, где C
– молярная концентрация раствора. Вместо концентрации C
по оси абсцисс можно откладывать значение объема аликвоты (V
) стандартного раствора или же массы вещества в нем. Вид графика при этом должен сохраняться.
Толщину слоя анализируемого раствора при фотометрировании можно варьировать от 0,2 до 5см
. Оптимальным считается такое значение l
, при котором диапазон изменения оптической плотности соответствует интервалу 0,1< A
< 0,8; при этом относительная погрешность измерений минимальна и составляет 0,5 −1,0%. Наиболее часто используются кюветы с толщиной слоя 1см.
Определение ионов меди (II
) основано на образовании ими комплексного соединения с аммиаком-тетрааммиаката меди с интенсивно голубой окраской по реакции:
Cu
2
+
+ 4NH
4
+
→[Cu
(NH
3
)4
]2
+
.
Этот метод применим для определения содержания меди в сплавах, лаках, электролитах и объектах окружающей среды.
3 Приборы и реактивы
ФЭК
− 56М
с 3 кюветами толщиной 10 мм
; мерные колбы на 25 мл
– 6 шт.; градуированная пипетка на 10 мл
; мерный цилиндр на 10 мл
; фильтровальная бумага; матерчатые салфетки.
Жидкие реактивы: стандартный раствор CuSO
4
с концентрацией
Т
= 6,5 мг
/см
3
; раствор аммиака (NH
4
OH
) с w
=12,5%; дистиллированная вода.
4
Указания по технике безопасности
Смотрите на стр. 13 (лабораторная работа 18).
5
Методика и порядок выполнения работы
1. Подготовка прибора
Для определения содержания ионов меди (II
) в растворе используется фотоэлектроколориметр ФЭК
− 56М
, устройство которого представлено на рис. 19.1.
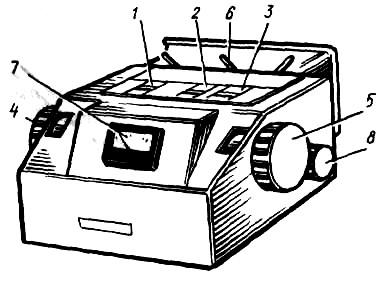
Рисунок 19.1 – Общий вид фотоэлектроколориметра-нефелометра ФЭК
− 56М
:
1, 2 и 3 – кюветодержатели; 4 – левый отчетный барабан; 5 – правый отчетный барабан; 6 – рукоятка переключения шторки; 7 – микроамперметр; 8 – рукоятка перемещения кювет
Прибор перед измерениями должен быть прогрет не менее 10 −15 минут для стабилизации работы лампы. Рукояткой устанавливают необходимый светофильтр №
8 (l
= 610 − 620 нм
).
Параллельно с прогревом прибора готовят серию стандартных растворов и пробу анализируемого вещества.
2. Приготовление серии стандартных растворов
Приготовьте серию стандартных растворов, для чего в мерные колбы
(№
1− 5) на 25 мл
отмерить по 5 мл
воды и последовательно пипеткой внести: 4;5;6;8 и 10 мл
стандартного раствора CuSO
4
соответственно. С помощью мерного цилиндра на 10 мл
или пробирки добавьте по 5 мл
аммиака и доведите объем раствора в каждой колбе дистиллированной водой до метки, добавляя последние порции воды по каплям. Раствор тщательно перемешайте.
3. Построение градуировочного графика
Измерьте оптическую плотность стандартных растворов CuSO
4
, используя в качестве раствора сравнения дистиллированную воду. Измерения начните с раствора наименьшей концентрации (№
1), а затем последовательно фотометрируйте остальные.
Кюветы необходимо тщательно промыть дистиллированной водой и удалить с поверхности все капли фильтровальной бумагой или салфеткой, измерительную кювету промыть дополнительно 2 − 3 раза фотометрируемым раствором. При замене пробы кювету следует ополоснуть 2 − 3 раза новым раствором
.
Внимание!
Кюветы можно держать пальцами только за боковые стенки во избежание загрязнения рабочих поверхностей жировой пленкой.
В кюветодержатели 1 и 2 помещают кюветы с растворителем (рис. 19.1), а кювету с испытуемым раствором – в кюветодержатель 3. При этом кюветы 2 и 3 должны находиться в крайнем левом положении.
Закрыв шторку кюветной камеры, устанавливают показания левого 4 и правого 5 барабанов на «0» по красной шкале. При помощи рукоятки «нуль» устанавливают стрелку микроамперметра на «0».
Рукояткой 6 открывают шторку и отклонившуюся стрелку микроамперметра 7 устанавливают на «0» при помощи левого барабана.
Закрывают шторку и рукояткой перемещения кювет 8 переводят кюветы 2 и 3 в крайнее правое положение.
Снова рукояткой 6 открывают шторку. Стрелку микроамперметра устанавливают на «0» с помощью правого барабана и закрывают шторку.
Отсчитывают значение абсорбционности по красной шкале правого барабана. По черной шкале правого барабана можно сделать отсчет коэффициента светопропускания.
Внимание!
При замене испытуемого раствора, прежде чем открыть крышку кюветного отсека, убедитесь в том, что шторка закрыта.
Запись данных опыта.
Постройте график зависимости оптической плотности A
от объема аликвоты раствора V
.
Результаты измерений и расчеты занесите в отчет и таблицу 19.1.
Таблица 19.1 – Данные для построения градуировочного графика
| №
колбы
|
Объем аликвоты стандартного
раствора меди,
V
,см
3
|
Оптическая плотность растворов,
A
|
масса ионов
Cu
2
+
в растворе m
, мкг
|
| 1
|
| 2
|
| 3
|
| 4
|
| 5
|
| 6[1]
|
4. Определение содержания ионов меди в контрольном образце
3.1. Получите у лаборанта в мерную колбу на 25 см
3
(№
6), содержащую 5см
3
дистиллированной воды аликвоту контрольного раствора меди, и подготовьте ее к фотометрированию как описано в пункте 2.
3.2. Определите значение оптической плотности контрольного раствора с помощью ФЭК
− 56М
и занесите его в табл. 19.1. По градуировочному графику найдите объем аликвоты.
Запись данных опыта.
Рассчитайте массу ионов меди в контрольном растворе при концентрации Cu
2
+
6,5 мг
/см
3
. Точность полученного результата проверьте у лаборанта или преподавателя.
6 Содержание отчета и его форма
Каждый студент должен иметь лабораторный журнал, который является документом, отражающим всю его работу.
Все наблюдения и выводы по экспериментальной работе, проделанной в лаборатории, студент заносит в лабораторный журнал непосредственно после ее выполнения в виде отчета.
Рекомендуется следующая схема записи:
1. Дата.
2. Номер и название лабораторной работы.
3. Цель работы.
4. Название и описание хода опытов.
5. Наблюдения.
6. Уравнения химических реакций.
7. Выводы и расчеты (формулы, таблицы, графики).
7 Вопросы для защиты работы
1. Классификация спектральных и оптических методов анализа.
Чувствительность методов, определяемые вещества.
2. Понятие спектра вещества, виды спектров.
3. Виды взаимодействия электромагнитного излучения с веществом.
4. Основные оптические характеристики веществ (интенсивность светового потока, относительное пропускание или поглощение света, молярный коэффициент светопоглощения, оптическая плотность) и их суть.
5. Основной закон светопоглощения и условия его применения. Свойства оптической плотности.
6. Чувствительность и точность фотоколориметрических определений. Выбор оптимальных условий фотометрирования.
7. Основные стадии фотометрического анализа.
8. Требования, предъявляемые к фотометрируемым веществам.
9. Холостая проба (раствор), методика его приготовления и назначение.
10. Основные методы определений содержания вещества (метод градуировочных графиков, метод добавок, метод сравнения и др.).
11. Устройство и принципиальная схема фотоэлектроколориметра ФЭК
− 56М
.
12. Светофильтры (определение, назначение, правило подбора по спектрам пропускания и цвету раствора).
13. Способы регистрации оптической плотности. Фотоэлемент, его устройство и принцип работы.
14. Молярный коэффициент светопоглощения (e
), его физический и графический смысл. Факторы, влияющие на величину e
(длина волны проходящего света, концентрация раствора, температура среды).
15. Условия фотометрирования растворов, содержащих несколько компонентов без их предварительного разделения.
16. Приведите примеры использования в фотометрическом анализе для получения окрашенных соединений таких химических реакций, как комплексообразование, ОВР, реакций осаждения, реакций образования или разрушения органических соединений.
17. Будет ли сохраняться линейной зависимость А
от С
, если:
а) состав анализируемого раствора при разбавлении не изменяется; б) при разбавлении раствора протекает гидролиз определяемого вещества; в) при разбавлении раствора изменяется степень диссоциации определяемого вещества; г) разбавление раствора приводит к изменению кислотности среды за счет смещения равновесия в растворе; д) при разбавлении показатель преломления раствора существенно изменяется.
8 Литература
[1, 2, 3, 4, 5, 6, 8, 10, 16, 17]
ЛАБОРАТОРНАЯ РАБОТА 20
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛАКТОЗЫ (САХАРОЗЫ) МЕТОДОМ РЕФРАКТОМЕТРИИ
1 Цель и содержание
Изучить принцип, устройство и методику работы на рефрактометре. Научиться определять плотности растворов с помощью пикнометра, строить градуировочные графики по экспериментальным данным и применять их в практических расчетах. Освоить метод наименьших квадратов и определения состава 3-х компонентных систем по номограмме.
2 Теоретическое обоснование
Метод рефрактометрии основан на измерении относительного показателя преломления света n
при прохождении луча через границу раздела прозрачных однородных сред, который является индивидуальной характеристикой вещества. Достоинство рефрактометрии в высокой точности метода 0,01− 0,1% и быстроте измерений при малом расходе растворов.
Различают абсолютный N
и относительный n
показатели преломления света. Под абсолютным показателем преломления понимают отношение скорости света в вакууме V
0
к скорости света в данной среде V
ср
: N
=V
0
/V
cp
. Относительным показателем преломления называют отношение скорости света в воздухе к скорости света в данной среде, то есть n
=V
B
/V
cp
.
Он зависит от природы вещества и растворителя, температуры среды, длины волны света.
Значение показателя преломления всегда больше 1, так как скорость света в вакууме (V
0
= 3⋅108
м
/с
) значительно выше, чем в плотной среде.
Для решения большинства практических задач аналитической химии достаточно точности измерения относительного показателя преломления и в расчеты не требуется вводить поправку на воздух: N
0
=1,00027 (абсолютный показатель преломления воздуха). В этом случае считают, что справедливо приближение: N
=1,00027 ⋅ n
≈ n
,
где n
− показатель преломления среды.
На рисунке 20.1 показано изменение хода луча света на границе раздела «воздух – среда», где α – угол падения луча света, β – угол преломления.
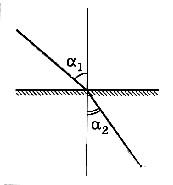
Рисунок 20.1 – Соотношение углов падения (a
1
) и преломления (a
2
) света для системы «воздух – среда»
Угол падения и преломления света связаны между собой соотношением: n
= sina
1
/sina
2
(закон Снеллиуса). Если вместо воздуха взять вторую среду с показателем преломления n
2
, то закон Снеллиуса можно преобразовать к виду: n
1
/n
2
= sina
2
/sina
1
, где n
1
− показатель преломления первой среды. Формулу Снеллиуса можно упростить, создавая такие условия, при которых угол a
1
= 900
, тогда n
1
= n
2
sina
2
− основная формула рефрактометрии.
В аналитических целях определяют зависимость показателя преломления n
от концентрации раствора C
при естественном освещении. При решении более сложных задач может использоваться монохроматическое излучение. Для рефрактометрии пригодны растворы, которые слабо окрашены, устойчивы на воздухе, имеют относительно высокую концентрацию (≈1%). Потерями излучения на отражение и поглощение света пренебрегают. Чтобы построить градуировочный график, необходимо подобрать концентрации стандартных растворов таким образом, чтобы область значений охватывала все возможные диапазоны концентраций исследуемых растворов, а зависимость в координатах n
− C
была линейна. При построении графика вместо концентрации C
по оси абсцисс можно откладывать также значения объемов аликвот (V
) стандартного раствора или же массу вещества. Вид графика при этом должен сохраняться.
Один из способов обработки данных при линейной зависимости параметров – метод наименьших квадратов (МНК)
, который позволяет рассчитать начальную координату параметра (n
0
) и тангенс угла наклона графика к оси абсцисс k
.
Для определения состава трех компонентных взаимосвязанных систем используют специальные графики – номограммы, которые получены экспериментально путем многократного измерения показателя преломления стандартных растворов различной концентрации. Вид номограммы зависит от особенностей изучаемого объекта и может быть представлен как линейными графиками, так и в виде области на координатной плоскости
(рис.20.2).

Рисунок 20.2 – Вид номограммы для расчета состава тройной системы:
вода – этанол – сахароза
При определении состава предварительно измеряют заданные параметры испытуемого раствора, по которым рассчитывают координаты точки на номограмме.
3 Приборы и реактивы
Рефрактометр марки ИРФ
− 420 или аналогичный; мерные колбы на
25 мл
– 5 шт.; градуированная пипетка на 10 мл
; капельница с дистиллированной водой; мягкая ткань и фильтровальная бумага.
Жидкие реактивы: стандартный раствор лактозы (С
12
Н
22
О
11
⋅ Н
2
О
) с концентрацией 0,5 моль
/ л
.
4
Указания по технике безопасности
Смотрите на стр. 13 (лабораторная работа 18).
5
Методика и порядок выполнения работы
1. Подготовка рефрактометра к работе
1.1. Для проверки правильности работы рефрактометра запишите температуру окружающей среды по контрольному термометру и рассчитайте теоретическое значение показателя преломления воды при данной температуре по формуле 20.1:
n
t
= n
20
+ (20 − t
) ⋅0,0002. (20.1)
1.2. Поместите на измерительную призму пипеткой немного дистиллированной воды и измерьте показатель ее преломления при данной температуре. Полученное экспериментально значение показателя преломления воды не должно отличаться от вычисленного по формуле (20.1) более, чем на 2⋅10−
4
. Если расхождение больше, то прибор требует настройки и им пользоваться нельзя.
Запись данных опыта.
Расчеты, теоретическое и экспериментальное значение n
t
приведите в отчете.
2. Приготовление серии стандартных растворов
Приготовьте серию стандартных растворов методом разведения концентрированного раствора. Для этого возьмите пять мерных колбочек объемом 25 мл
. С помощью пипетки с делениями на 10 мл
отмерьте в 1− ю
колбу – 2, во 2 − ю
– 4, в 3− ю
– 6; в 4 − ю
– 8 и в 5 − ю
– 10 мл
раствора лактозы с концентрацией 0,5 моль
/ л
. Объем раствора в каждой колбочке доведите до 25 мл
дистиллированной водой и тщательно перемешайте.
Иногда в растворе лактозы может на дне появляться незначительный осадок, который не мешает измерениям.
3. Построение градуировочного графика
3.1. Протрите призмы рефрактометра мягкой тканью или осушите фильтровальной бумагой. Проведите измерение показателя преломления стандартных растворов лактозы, начиная с наименьшей концентрации (№
1).
Для этого нанесите на измерительную призму пипеткой небольшое количество анализируемого раствора и промойте им призму, а затем повторно нанесите измеряемый раствор и определите показатель его преломления. Каждый замер проводите 3 раза.
Запись данных опыта.
Полученные данные занесите в таблицу 20.1 и вычислите среднее значение показателя преломления.
3.2. Рассчитайте концентрацию стандартных растворов лактозы по формуле (20.2) и занесите в таблицу 20.1.
С
ст
⋅V
ст
= С
р
⋅V
р
, (20.2)
где С
ст
– концентрация стандартного раствора лактозы; V
ст
– объем аликвоты стандартного раствора, С
р
– концентрация лактозы в растворе после разведения; V
p
– объем полученного раствора лактозы (25 мл
).
Запись данных опыта.
По полученным данным постройте градуировочный график: n
= f
(C
).
Таблица 20.1 – Результаты измерения показателя преломления стандартных растворов лактозы
| №
колбы
|
Объем раствора лактозы,
V
, мл
|
Концентрация раствора,
С
М
, моль
/ л
|
Значения показателя преломления
|
Масса лактозы в контрольном растворе m
, г
, рассчитанная
|
| n
1
|
n
2
|
n
3
|
n
ср
|
по графику
|
методом МНК
|
| 1
|
| 2
|
| 3
|
| 4
|
| 5
|
| 6[2]
|
4. Определение содержания лактозы в контрольном растворе по градуировочному графику и по методу МНК
4.1. Получите аликвоту контрольного раствора лактозы у лаборанта в мерную колбу на 25 мл
, доведите объем до метки и перемешайте.
Определите показатель преломления раствора лактозы с неизвестной концентрацией и запишите его значение в таблицу 20.1.
4.2. Используя полученный ранее градуировочный график, определите по нему концентрацию растворы лактозы в контрольном образце и рассчитайте массу лактозы в контрольной пробе.
4.3. Проведите расчет параметров n
0
, k
и C
для контрольного раствора с использованием метода МНК, учитывая, что зависимость показателя преломления раствора лактозы от концентрации линейна и описывается формулой 20.3:
n
= n
0
+ kC
, (20.3) где n
– измеренный показатель преломления исследуемого раствора; n
0
– показатель преломления растворителя (точка пересечения графика с осью ординат); k
= tg
a
, где a
– угол наклона градуировочного графика к оси абсцисс; C
– молярная концентрация определяемого вещества в контрольном растворе, моль
/ л
.
Пусть m
– число измерений показателя преломления стандартных растворов, тогда по формулам (20.4) и (20.5) можно вычислить значения n
0
и k
:
∑m n
i
− k
⋅∑m c
i
n
0
=  i
=
1 i
=
1
, (20.4) i
=
1 i
=
1
, (20.4)
m
m
⋅ ∑m c
in
i
− ∑m c
i
⋅∑m n
i
 k
= k
=  i
=
1 i
=
1 i
=
1
2
. (20.5) m
⋅ ∑im c
2 − ∑im
=1 c
i
i
=
1 i
=
1 i
=
1
2
. (20.5) m
⋅ ∑im c
2 − ∑im
=1 c
i
При вычислениях удобно записывать промежуточные результаты в виде таблицы 20.2.
Таблица 20.2 – Данные для расчета параметров по методу МНК
| номер опыта →
|
1
|
2
|
3
|
4
|
5
|
полученное
значение суммы
|
| параметр ↓
|
| концентрация лактозы C
, моль
/ л
|
∑c
i
=
|
| показатель преломления, n
cp
, i
|
∑n
i
=
|
| произведение n
i
⋅c
i
|
∑(c
i
⋅n
i
) =
|
| квадрат концентрации, c
i
2
|

|
4.4. По методу МНК, используя экспериментальные значения из таблицы 20.1, вычислите значения n
0
и k
,
а по формуле (20.3) рассчитайте концентрацию лактозы в контрольной пробе и найдите массу вещества в пробе. Сравните результаты вычисления массы лактозы, полученные двумя методами.
Запись данных опыта.
Все данные занесите в таблицы 20.1 и 20.2.
6 Содержание отчета и его форма
Каждый студент должен иметь лабораторный журнал, который является документом, отражающим всю его работу.
Все наблюдения и выводы по экспериментальной работе, проделанной в лаборатории, студент заносит в лабораторный журнал непосредственно после ее выполнения в виде отчета.
Рекомендуется следующая схема записи:
1. Дата.
2. Номер и название лабораторной работы.
3. Цель работы.
4. Название и описание хода опытов.
5. Наблюдения.
6. Уравнения химических реакций.
7. Выводы и расчеты (формулы, таблицы, графики).
7 Вопросы для защиты работы
1. Дайте определение абсолютного и относительного показателей преломления. Почему в выполненной работе не учитывалась поправка на воздух?
2. Способ определения показателя преломления среды, свойства показателя преломления.
3. Связь показателя преломления с углами падения и преломления (закон Снеллиуса), понятие предельного или критического угла. Условие полного внутреннего отражения.
4. Что понимают под дисперсией света и как ее определяют? Как можно по величине дисперсии сравнить преломляющие свойства среды?
5. Молярная рефракция: определение и расчет, факторы, влияющие на ее значение. Уравнение Лорентца-Лоренца.
6. На чем основан метод рефрактометрического анализа? Область применения метода (укажите, какие параметры среды и свойства веществ можно рассчитывать по показателю преломления).
7. Требования к растворам, применяемым в рефрактометрии.
8. Факторы, влияющие на значение показателя преломления.
9. Устройство рефрактометра и принцип его работы. Как можно проверить точность показаний прибора?
10. Применение рефрактометрии в пищевой промышленности, анализе органических и неорганических веществ.
11. Сущность других оптических методов (фотоколориметрия, нефелометрия, турбидиметрия, поляриметрия).
8 Литература
[1, 2, 3, 4, 5, 6, 8, 10, 16, 17]
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
ЛАБОРАТОРНАЯ РАБОТА 21
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КАРБОНАТА И ГИДРОКАРБОНАТА В СМЕСИ МЕТОДОМ ПРЯМОГО ПОТЕНЦИОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
1 Цель и содержание
Освоить навыки потенциометрического титрования по кислотноосновному механизму; выполнить дифференциальное определение смеси карбоната и гидрокарбоната натрия.
2 Теоретическое обоснование
Потенциометрическое титрование
относится к косвенным методам анализа, основано на установлении точки эквивалентности по резкому изменению потенциала индикаторного электрода при титровании. Титрант добавляют точно известными порциями и записывают показания прибора. По результатам титрования строят интегральную и (или) дифференциальную кривые титрования (рис. 21.1). По графикам находят точку эквивалентности и выполняют расчет.
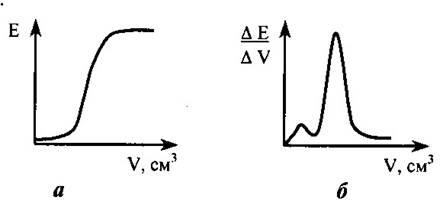
Рисунок 21.1 – Интегральная (а
) и дифференциальная (б
) кривые титрования смеси двух веществ
Выбор индикаторных электродов зависит от природы определяемых ионов и типа химической реакции. В таблице 21.1 приведены некоторые варианты выбора электродов при потенциометрическом титровании. В качестве электродов сравнения, как правило, применяют насыщенные электроды II
рода.
Таблица 21.1 – Виды потенциометрического титрования
| Тип реакции
|
Измеряемая величина
|
Электроды
|
Определяемые вещества
|
| Индикаторный
|
Сравнения
|
| Протолитометрия
|
рН
|
рН
-стеклянный,
хингидронный
|
Насыщенные электроды II
рода
(хлоридсеребряный,
каломель- ный)
|
Кислоты, основания, соли
|
| Редоксиметрия
|
Е
|
Индифферентные
I
рода
(платиновый)
|
Окислители, восстановители
|
| Комплексо нометрия
|
рМе
|
Me
-селективные
|
Me
n
+
; n
>1
|
| Осаждение
|
pAg
, pCl
,
pI
, pBr
|
Ионоселективные; ненасыщенные II
рода; серебряный
|
Ионы, образующие осадки
|
Основными достоинствами метода потенциометрического титрования являются высокая точность и возможность проводить определения в разбавленных растворах, в мутных и окрашенных средах, с использованием неводных растворителей, а также определять несколько веществ в одном растворе без предварительного разделения.
К недостаткам можно отнести не всегда быстрое установление потенциала после добавления титранта и необходимость во многих случаях проводить при титровании большое количество отсчётов.
Установка для титрования.
Для потенциометрического титрования необходимы индикаторный электрод, электрод сравнения, прибор для измерения разности потенциалов между ними, ячейка, бюретка с раствором титранта (рис. 21.2).
Электроды для измерения рН.
В качестве индикаторного применяют стеклянный электрод, который относится к мембранным ионоселективным электродам. Мембрана электрода изготовлена из тонкого стекла, поэтому при работе следует проявлять осторожность!
Для измерения потенциала индикаторного электрода необходим стандартный электрод сравнения. В качестве электрода сравнения применяют насыщенный хлоридсеребряный электрод.
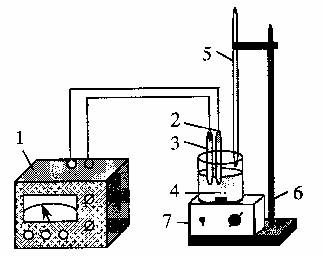
Рисунок 21.2 – Установка для потенциометрического титрования:
1 – рН-метр; 2 – стеклянный электрод; 3 – электрод сравнения (хлорид серебряный); 4 – ячейка с раствором; 5 – бюретка; 6 – штатив; 7 – магнитная мешалка
Устройство универсального иономера ЭВ-74
На передней стенке прибора (рис. 21.3) расположен гальванометр 1, по показаниям стрелки 2 которого определяется рН
или мВ
измеряемой цепи.
Зеркальная дуга 3 предназначена для повышения точности отсчёта показаний. Для этого глаз должен находиться в положении, при котором стрелка и её отражение в зеркале совмещаются.
В правом верхнем углу передней панели расположены тумблер включения 4 и глазок индикации включения 5.
Кнопка «Анионы/Катионы (±)», когда она не нажата, позволяет производить измерение концентрации анионов (или снимать положительные значения потенциалов), а когда нажата – концентрации катионов (отрицательные значения потенциалов).
Кнопка « Х
′/ Х
′′» – используется при измерении концентрации однозарядных (не нажата) и двухзарядных (нажата) ионов.
Кнопкой «mB
» прибор включается в режим милливольтметра, кнопкой « pX
» – в режим иономера и pH
-метра. При нажатой кнопке «t
o
» производится ручная установка температуры раствора.
Кнопки правого ряда включают соответствующие диапазоны величины pX
в режиме иономера. Ручки оперативного управления 7 предназначены для настройки прибора на данную электродную систему и температуру раствора (для предотвращения случайного проворачивания закрыты прозрачными плёнками).

Рисунок 21.3 – Прибор «Иономер универсальный ЭВ
− 74»:
1 − гальванометр; 2 − стрелка гальванометра; 3 − зеркало; 4 − включение прибора; 5 − глазок (лампочка) включения прибора; 6 − клавиши рода работ и диапазонов измерений;
7−ручки управления прибором; 8 − винт установки стрелки на «нуль»
Анализ содержания карбоната и гидрокарбоната натрия
при их совместном присутствии в растворе основан на определении в ходе потенциометрического титрования двух скачков на кривой титрования. В качестве титранта применяется соляная кислота. Первый скачёк соответствует реакции:
Na
2
CO
3
+ HCl
→ NaHCO
3
+ NaCl
, (21.1)
а второй:
NaHCO
3
+ HCl
→ CO
2
+ NaCl
+ H
2
O
, (21.2)
причём в реакцию (21.2) вступает как гидрокарбонат натрия, присутствующий в исходном растворе, так и тот, который образовался в ходе реакции (21.1) из Na
2
CO
3
.
Обозначим искомые количества эквивалентов как n
э
(NaHCO
3
) и n
э
(Na
3
CO
3
). Тогда количество эквивалентов HCl
, пошедшее на реакцию
(21.1):
n
э
′(HCl
) = n
э
(Na
2
CO
3
) = n
э
′
(NaHCO
3
), (21.3)
где n
э
′(NaHCO
3
) – количество эквивалентов гидрокарбоната образовавшегося из Na
2
CO
3
оттитрованного наполовину.
Количество эквивалентов HCl
, пошедшее на реакцию (21.2):
n
э
″(HCl
) = n
э
(NaHCO
3
) + n
э
″
(NaHCO
3
). (21.4)
При подстановке выражения (21.3) в уравнение (21.4) получим уравнение для расчета количества эквивалентов исходного гидрокарбоната натрия:
n
э
(NaHCO
3
) = n
э
″
(HCl
) − n
э
′
(HCl
). (21.5)
Если C
н
(HCl
) – концентрация титранта, V
ал
– объём аликвоты анализируемой смеси, а V
′ и V
′′ – объёмы титранта, израсходованные соответственно к первому и второму скачку, то используя равенство (21.3), можно рассчитать концентрацию карбоната натрия по формуле:
C
н
(Na
2
CO
3
) = C
н
(HCl
) ⋅2V
′/V
ал
. (21.6)
Для того чтобы вывести формулу для концентрации гидрокарбоната натрия, необходимо учесть, что после достижения точки эквивалентности в реакции (21.1) уровень титранта в бюретке не выводится на нуль, а начинает расходоваться на реакцию (21.2) от значения V
′. Следовательно, объём титранта соответствующий n
э
″(HCl
) равен разности V
′′ и 2V
′.
В итоге из уравнения (21.6) получим:
C
н
(NaHCO
3
) = C
н
(HCl
)⋅(V
′′ − 2V
′)/V
ал
. (21.7)
3 Приборы и реактивы
Иономер универсальный ЭВ
− 74 со стеклянным электродом; стакан с
1 моль
/ л
раствором HCl
на 150 мл
; мерная колба на 100 мл
– 1 шт.; пипетка Мора на 10 мл
; промывалка с дистиллированной водой; магнитная мешалка; бюретка на 25 мл
; термометр до 50o
С
; фильтровальная бумага.
Жидкие реактивы: контрольный раствор смеси Na
2
CO
3
и NaHCO
3
.
4
Указания по технике безопасности
Смотрите на стр. 13 (лабораторная работа 18).
5
Методика и порядок выполнения работы
1. Подготовка к работе иономера ЭВ
− 74
1.1. Ознакомьтесь внимательно с устройством и порядком работы на универсальном иономере ЭВ
− 74.
1.2. Включите прибор для прогревания на 15− 20 минут (кнопка «t
o
» должна быть в нажатом состоянии, а остальные выключены).
1.3. Раствор HCl
, в котором хранятся электроды, из стакана перелить в отдельную колбу, а электроды промыть несколько раз дистиллированной водой с использованием промывалки над стаканом и осушить фильтровальной бумагой.
2. Подготовка установки для титрования
2.1. Получите у лаборанта в мерную колбу на 100 мл
аликвоту раствора, содержащую смесь карбоната и гидрокарбоната натрия, доведите его до метки дистиллированной водой и перемешайте.
2.2. Заполнить бюретку для титрования раствором HCl
(точную концентрацию указывает лаборант), предварительно её промыв этим раствором и удалив воздушный пузырек из носика.
2.3. Отобрать аликвоту анализируемого раствора пипеткой Мора на 10,00 мл и поместить ее в стакан для титрования.
2.4. В стакан добавить дистиллированной воды так, чтобы электроды были погружены в раствор на 2 см
, туда же опустить магнит и включить магнитную мешалку. Расстояние от электродов до магнита примерно 0,5 −1см
.
3. Проведение ориентировочного (грубого) титрования
3.1. Нажать на приборе кнопки (+/−) и «−1−19», «Анионы/ катионы
», « рХ
», при этом кнопка «t
o
» должна отжаться. Отметить по шкале гальванометра −1−19 значение рН исходного раствора (V
титранта равен 0 мл
) и занести в таблицу результатов ориентировочного титрования
(табл.7).
3.2. Провести грубое титрование, добавляя титрант по 1 мл
. Его объём и значение рН
(необходимо подождать пока стрелка установится) записывают в табл. 21.2. Титрование проводят до обнаружения двух скачков по резкому изменению значений pH. Рассчитать ∆рН
1
= рН
2
− рН
1
и т. д.
3.3. Закончив титрование, нажать кнопку «t
o
», отключить остальные кнопки на приборе и выключить магнитную мешалку. Раствор из стакана вылить, а электроды промыть дистиллированной водой.
Запись данных опыта.
По данным табл. 21.2 определяют интервалы
∆V
1
и ∆V
2
титранта, в течение которых протекают скачки, а так же найти графически значения V
1
и V
2
. Построить на миллиметровой бумаге интегральную кривую титрования.
Таблица 21.2 – Результаты ориентировочного (грубого) титрования
| №
|
Объём титранта HCl
, V
, мл
|
рН
|
∆рН
|
| 1
|
| 2
|
| 3
|
| …
|
4. Проведение точного титрования
4.1. Подготовить установку к точному титрованию согласно п. 2 и 3 (перед промыванием стакана не забудьте извлечь магнит).
4.2. Не добавляя титранта, определить значение рН
по шкале
«−1−19» и перейти на точный диапазон, нажав одну из кнопок: «−1− 4», «4 −9», «9 −14», «14 −19». Если используется диапазон «−1− 4», то значения рН
определяются по шкале гальванометра (−1− 4). В остальных случаях рН
определяют по шкале «
0 − 5»
, для чего к показаниям шкалы
«
0 − 5»
необходимо прибавить число, соответствующее началу выбранного диапазона. Например, если выбран диапазон 9 −14, а показание по шкале 0 − 5 равно 2,7, то значение рН
составит: рН
= 9 + 2,7 =11,7.
4.3. Записать точное значение рН
при V
= 0 и вновь перейти на диапазон «−1−19». Быстро слить раствор HCl
, не доходя примерно 0,5 − 0,8 мл
до начала первого скачка V
1
. Записать значение рН
.
4.4. Перейти на точный диапазон, как показано в п.4.2, записать точное значение рН
и провести точное титрование в интервале ∆V
1
, добавляя титрант по 0,2 мл
. Объём титранта и значение рН
записывать в табл. 21.3.
По окончании первого скачка вновь перейти на диапазон «−1−19». Конец скачка определяют в момент, когда ∆V
практически не меняется.
4.5. Быстро приливая титрант, оттитровать раствор до начала второго скачка V
2
, не доходя до него 0,5 − 0,8 мл
, и вновь перейти на точный диапазон измерения рН
. Провести точное титрование в интервале ∆V
2
, добавляя титрант по 0,2 мл
и записывая объем титранта и показания прибора в табл. 21.3.
4.6. Окончив титрование, перейдите к диапазону (−1−19) и нажмите кнопку «t
o
». Выключите прибор, промойте бюретку, электроды и стакан для титрования дистиллированной водой. Заполните стакан для хранения электродов раствором HCl
и опустите в него электроды.
Таблица 21.3 – Результаты точного титрования
| №
|
V
титранта, мл
|
рН
|
∆рН
|
∆рН
/ ∆V
|
| 1
|
| 2
|
| 3
|
| …
|
Запись данных опыта.
Рассчитайте значения ∆рН
и ∆рН
/∆V
и занесите их в табл. 21.3. По данным табл. 21.3 постройте на миллиметровой бумаге дифференциальную кривую титрования. Определить по экстремумам эквивалентные объёмы V
′ и V
′′. Рассчитайте по формулам (21.6) и (21.7) концентрации карбоната и гидрокарбоната натрия в растворе, а также их массы в пробе. Сравните значения V
1
и V
2
, полученные по интегральной и дифференциальной кривым, сделайте вывод.
6 Содержание отчета и его форма
Каждый студент должен иметь лабораторный журнал, который является документом, отражающим всю его работу.
Все наблюдения и выводы по экспериментальной работе, проделанной в лаборатории, студент заносит в лабораторный журнал непосредственно после ее выполнения.
Рекомендуется следующая схема записи:
1. Дата.
2. Номер и название лабораторной работы.
3. Цель работы.
4. Название и описание хода опытов.
5. Наблюдения.
6. Уравнения химических реакций.
7. Выводы и расчеты (формулы, таблицы, графики).
7 Вопросы для защиты работы
1. Какие методы объединяют в группу электрохимических методов анализа, какие параметры среды измеряются?
2. Устройство и принцип работы иономера ЭВ–74. Какие измерительные схемы применяются в потенциометрии?
3. В чем отличие прямой потенциометрии от потенциометрического титрования? Преимущества потенциометрического титрования по сравнению с традиционным.
4. Устройство и принцип работы измерительного электрода и электрода сравнения на примере стеклянного и хлоридсеребрянного. Какого рода эти электроды?
5. Требования к измерительному электроду и электроду сравнения. Факторы, влияющие на показание электродов.
6. Водородный электрод: устройство, принцип работы и назначение. Электрохимический ряд напряженности металлов.
7. Ионоселективные электроды и их назначение.
8. Газодинамические электроды: устройство и назначение.
9. Интегральная и дифференциальная кривые потенцио-
метрического титрования: построение и определение точки эквивалентности. В чем достоинство дифференциальной формы кривой титрования?
10. Какие вещества можно анализировать, используя метод потенциометрии, каким условиям они должны удовлетворять?
11. Кондуктометрия как метод анализа.
12. Виды кондуктометрических ячеек и их назначение.
8 Литература
[1, 3, 7, 13, 14, 15]
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Основная
1. Харитонов, Ю. Я. Аналитическая химия. Аналитика /
Ю. Я. Харитонов. – М. : Высш. шк., 2001. – Ч. 2 – 225 с.
2. Цитович, И. К. Курс аналитической химии.: учебник. 9-е изд., стер. / И. К. Цитович. – СПб. : Лань, 2007. – 496 с.
3. Основы аналитической химии / под ред. Ю.А.Золотова. – М. : Высш. шк., 1999. – Ч. 2 – 494 с.
4. Основы аналитической химии. Практическое руководство / под ред. Ю. А. Золотова. – М. : Высш. шк., 2001. – 463 с.
5. Коренман, Я. И. Практикум по аналитической химии. Анализ пищевых продуктов / Я. И. Коренман, Р. П. Лисицкая. – Воронеж : Воронеж.
гос. технол. акад., 2002. – 408 с.
6. Васильев, В. П. Практикум по аналитической химии /
В. П. Васильев, Р. П. Морозова, Л. А. Кочергина. – М. : Химия, 2000. – 328 с.
7. Бегишева, С. Ш. ЭУК «Аналитическая химия и ФХМА» /
С. Ш. Бегишева, О. А. Слепышева. – Ставрополь : СевКавГТУ, 2007. – Гл. 9.
8. Слепышева, О. А. ЭУК «Аналитическая химия и ФХМА» /
О. А. Слепышева. – Ставрополь : СевКавГТУ, 2007. – Гл. 6, 8.
9. Слепышева, О. А. ЭУК «Аналитическая химия и ФХМА» / О. А. Слепышева. – Ставрополь : СевКавГТУ, 2007. – Гл. 7.
Дополнительная
10. Дорохова, Е. Н. Аналитическая химия. Физико-химические методы анализа / Е. Н. Дорохова, Г. В. Прохорова. – М.: Высш. шк.,1991. – 225 с.
11. Дорохова, Е. Н. Задачи и вопросы по аналитической химии / Е. Н. Дорохова, Г. В. Прохорова. – М. : Мир, 2001. – 268 с.
12. Коренман, Я. И. Задачник по аналитической химии. Титриметрические методы анализа / Я. И. Коренман, П. Т. Суханов, С. П. Калинкина. – Воронеж : Воронеж. гос. технол. акад., 2001. – 336 с.
13. Лабораторное руководство по хроматографии и смежным методам / под ред. О. Микеш и др. – М. : Мир, 1982. – Ч. 2. – 215 с.
14. Практикум по физико-химическим методам анализа / под ред. О. М. Петрухина. – М. : Химия, 1987.
15. Столяров, Б. В. Руководство к практическим работам по газовой хроматографии / Б. В. Столяров, И. М. Савинов, А. Г. Виттенберг. – Ленинград : Химия, 1978. – 288 с.
16. Лурье, Ю. Ю. Справочник по аналитической химии /
Ю. Ю. Лурье. – М. : Химия, 1989. – 162 с.
17. Дерффель, К. Статистика в аналитической химии / К. Дерф- фель. – М. : Мир, 2001. – 267 с.
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
к выполнению лабораторных работ
по дисциплине «Аналитическая химия и
физико-химические методы анализа»
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
для студентов специальностей
240901 «Биотехнология»; 240902 «Пищевая биотехнология»;
260202 «Технология хлеба, кондитерских и макаронных изделий»;
260301 «Технология мяса и мясных продуктов»; 260303 «Технология молока и молочных продуктов»; 240403 «Химическая технология природных
энергоносителей и углеродных материалов»; 240306 «Химическая
технология монокристаллов, материалов и изделий электронной техники»; 240100 «Химическая технология и биотехнология – бакалавриат»
Составители: О. А. Слепышева, канд. хим. наук, доцент,
М. В. Грицаева, ассистент
Редактор: Л. Д. Бородастова
Подписано в печать 29.12.2008 г.
Формат 60х84 1/16. Усл. п. л. – 3,0. Уч.-изд. л. – 2,5
Бумага газетная Печать офсетная. Заказ Тираж 30 экз.
ГОУВПО «Северо-Кавказский государственный технический университет» 355028, г. Ставрополь, пр. Кулакова, 2.
Издательство «Северо-Кавказского государственного технического университета»
Отпечатано в типографии СевКавГТУ
[1]
Колба №6 содержит пробу контрольного раствора, данные для которого получают с помощью градуировочного графика.
[2]
Колба №6 содержит контрольную пробу раствора, данные для которой определяют по градуировочному графику.
|