| ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
ДАЛЬНЕВОСТОЧНЫЙ ГОСУДАРСТВЕННЫЙ ТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ
(ДВПИ им. В.В. Куйбышева)
АРСЕНЬЕВСКИЙ ТЕХНОЛОГИЧЕСКИЙ ИНСТИТУТ (филиал) ДВГТУ
Г.Г. ДЁМИЧ
Х И М И Я
(конспект лекций)
Рекомендовано Ученым советом
Арсеньевского технологического института в качестве учебного пособия
для студентов высших учебных заведений, обучающихся специальностям
151001 Технология машиностроения
140211 Электроснабжение
080502 Экономика и управление на предприятии (в машиностроении)
Арсеньев 2007
ОГЛАВЛЕНИЕ
ОГЛАВЛЕНИЕ .................................................................................................................... 1
ПРЕДИСЛОВИЕ ................................................................................................................. 7
ВВЕДЕНИЕ ....................................................................................................................... 11
Химия как раздел естествознания
....................................................................................................................... 11
Основные задачи современной химии
.............................................................................................................. 12
ЛЕКЦИЯ 1. ХИМИЧЕСКИЕ СИСТЕМЫ .......................................................................... 14
1.1.
Вещество как система
................................................................................................................................ 14
1.2.
Концептуальный уровень
........................................................................................................................ 15
1.3. Субатомный уровень
...................................................................................................................................... 22
1.4. Квантовый уровень
......................................................................................................................................... 28
1.5. Проблема строения вещества
...................................................................................................................... 40
1.6. Вещество на макроуровне
.............................................................................................................................. 43
1.7. Молекулярные системы
.................................................................................................................................. 47
1.8. Процессы, происходящие в химических системах
............................................................................... 54
1.9.
Вопросы и задания:
.................................................................................................................................... 57
ЛЕКЦИЯ 2. РАСТВОРЫ .................................................................................................. 60
2.1. Раствор как система
.......................................................................................................................................... 60
2.2 Растворение и растворимость
....................................................................................................................... 61
2.3 Способы выражения концентрации растворов
....................................................................................... 64
2.4. Электролитическая диссоциация
................................................................................................................ 67
2.5. Ионная сила раствора и коэффициент активности
.............................................................................. 70
2.6. Физические свойства растворов
................................................................................................................. 72
2.7. Вопросы и задания:
.......................................................................................................................................... 74
ЛЕКЦИЯ 3. ДИСПЕРСНЫЕ СИСТЕМЫ ......................................................................... 76
3.1. Классификация дисперсных систем
.......................................................................................................... 76
3.2. Коллоидные растворы
..................................................................................................................................... 78
3.3. Свойства коллоидных систем
...................................................................................................................... 81
3.4. Устойчивость коллоидных систем
.............................................................................................................. 84
3.5. Вопросы и задания:
.......................................................................................................................................... 87
ЛЕКЦИЯ 4. ЭЛЕКТРОХИМИЧЕСКИЕ СИСТЕМЫ ......................................................... 89
4.1. Гальванические элементы
............................................................................................................................. 89
4.2. Электролиз
........................................................................................................................................................... 99
4.3. Коррозия металлов
......................................................................................................................................... 106
4.4. Вопросы и задания:
........................................................................................................................................ 116
ЛЕКЦИЯ 5. КАТАЛИЗАТОРЫ И КАТАЛИТИЧЕСКИЕ СИСТЕМЫ ............................ 118
5.1. Механизм действия катализаторов
........................................................................................................... 119
5.2. Особенности каталитических систем
....................................................................................................... 123
5.3. Перспективы развития каталитических систем
................................................................................... 124
5.4. Вопросы и задания
......................................................................................................................................... 128
ЛЕКЦИЯ 6. ПОЛИМЕРЫ И ОЛИГОМЕРЫ ................................................................... 129
6.1. Органические соединения
............................................................................................................................ 129
6.2. Природные полимеры
................................................................................................................................... 132
6.3. Синтетические полимеры
............................................................................................................................. 140
6.4. Олигомеры
......................................................................................................................................................... 147
6.5. Вопросы и задания:
........................................................................................................................................ 148
ЛЕКЦИЯ 7. ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА И КИНЕТИКА .................................. 149
7.1. Химическая термодинамика
........................................................................................................................ 149
7.2. Законы термодинамики
................................................................................................................................. 150
7.3. Химическая кинетика
...................................................................................................................................... 154
7.4. Вопросы и задания
......................................................................................................................................... 159
ЛЕКЦИЯ 8. ЭНЕРГЕТИКА ХИМИЧЕСКИХ ПРОЦЕССОВ ........................................... 160
8.1. Термохимия
........................................................................................................................................................ 160
8.2 Теплота образования
...................................................................................................................................... 164
8.3 Энтальпия
............................................................................................................................................................ 166
8.4 Теплота сгорания
.............................................................................................................................................. 170
8.5. Вопросы и задания
......................................................................................................................................... 172
ЛЕКЦИЯ 9. ХИМИЧЕСКОЕ И ФАЗОВОЕ РАВНОВЕСИЕ .......................................... 173
9.1. Химическое равновесие
................................................................................................................................ 173
9.2. Константа равновесия
.................................................................................................................................... 173
9.3. Принцип Ле Шателье
...................................................................................................................................... 176
9.4. Фазовое равновесие
....................................................................................................................................... 179
9.5. Правило фаз
...................................................................................................................................................... 181
9.6. Диаграммы состояния
.................................................................................................................................... 182
9.7. Вопросы и задания:
........................................................................................................................................ 193
ЛЕКЦИЯ 10. СКОРОСТЬ РЕАКЦИИ И МЕТОДЫ ЕЕ РЕГУЛИРОВАНИЯ ................. 194
10.1 Природа реагирующих веществ
................................................................................................................ 194
10.2. Концентрация реагирующих веществ
.................................................................................................... 195
10.3. Температура реакционной среды
............................................................................................................ 195
10.4. Энергия активации
........................................................................................................................................ 197
10.5. Катализаторы
.................................................................................................................................................. 199
10.6. Вопросы и задания
....................................................................................................................................... 199
ЛЕКЦИЯ 11. КОЛЕБАТЕЛЬНЫЕ РЕАКЦИИ ................................................................ 201
11.1. Колебательные процессы в химии
......................................................................................................... 201
11.2. Реакция Белоусова-Жаботинского
.......................................................................................................... 204
11.3. Колебательные реакции и синергетика
................................................................................................. 207
11.4. Практическое применение колебательных процессов
................................................................... 211
11.5. Нанотехнологии
.............................................................................................................................................. 213
11.6. Вопросы и задания
....................................................................................................................................... 217
ЛЕКЦИЯ 12. РЕАКЦИОННАЯ СПОСОБНОСТЬ ВЕЩЕСТВ ...................................... 218
12.1. Химическое сродство
................................................................................................................................... 218
12.2. Энергия Гиббса
............................................................................................................................................... 221
12.3. Энергия Гельмгольца
................................................................................................................................... 222
12.4. Критерий возможности самопроизвольного протекания химических процессов
................ 224
12.5. Вопросы и задания
....................................................................................................................................... 227
ЛЕКЦИЯ 13. ХИМИЯ И ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ ...................... 230
13.1. Связь Периодической системы с квантовой химией
....................................................................... 231
13.2. Строение Периодической системы
......................................................................................................... 233
13.3. Электронные формулы элементов
......................................................................................................... 236
13.4. Краткий обзор свойств s-, p-, d- и f-элементов
................................................................................... 238
13.5. Вопросы и задания:
...................................................................................................................................... 257
ЛЕКЦИЯ 14. КИСЛОТНЫЕ, ОСНОВНЫЕ И ОКИСЛИТЕЛЬНОВОССТАНОВИТЕЛЬНЫЕ СВОЙСТВА ВЕЩЕСТВ .................................................... 260
14.1. Кислоты и основания
................................................................................................................................... 260
14.2. Водородный показатель
............................................................................................................................. 265
14.3. Гидролиз
........................................................................................................................................................... 269
14.4. Буферные растворы
.................................................................................................................................... 272
14.5. Окислительно-восстановительные процессы
................................................................................... 274
14.6. Вопросы и задания:
...................................................................................................................................... 281
ЛЕКЦИЯ 15. ХИМИЧЕСКАЯ СВЯЗЬ; КОМПЛИМЕНТАРНОСТЬ ............................... 284
15.1. Энергия ионизации и сродство к электрону
........................................................................................ 284
15.2. Валентность
..................................................................................................................................................... 286
15.3. Виды химической связи
.............................................................................................................................. 288
15.5. Комплементарность
...................................................................................................................................... 303
15.6. Гибридизация
.................................................................................................................................................. 306
15.7. Метод молекулярных орбиталей (ЛКАО МО)
...................................................................................... 307
15.8. Вопросы и задания
....................................................................................................................................... 309
ЛЕКЦИЯ 16. ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ ........................................................ 310
16.1. Качественный анализ
................................................................................................................................... 310
16.2. Количественный анализ
.............................................................................................................................. 323
16.3. Физические и физико-химические методы анализа
......................................................................... 327
16.4. Вопросы и задания:
...................................................................................................................................... 332
ЛЕКЦИЯ 17. ОСНОВЫ БИОХИМИЧЕСКИХ ПРОЦЕССОВ И ИХ ПРИМЕНЕНИЕ В ТЕХНОЛОГИЧЕСКОЙ ОТРАСЛИ ................................................................................. 334
17.1. Клетка – основа жизни
................................................................................................................................. 334
17.2. Метаболизм
...................................................................................................................................................... 337
17.3. Триплетный код и матричный синтез
.................................................................................................... 340
17.4. Биохимические процессы в технологической отрасли
................................................................... 346
17.5. Вопросы и задания
....................................................................................................................................... 356
ЛЕКЦИЯ 18. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕМБРАННЫХ ТЕХНОЛОГИЙ ............ 357
18.1. Строение биомембран
................................................................................................................................. 358
18.2. Процессы переноса
...................................................................................................................................... 360
18.3. Современные мембранные материалы и технологии
..................................................................... 364
18.3. Перспективы развития мембранных технологий
.............................................................................. 369
18.4. Вопросы и задания
....................................................................................................................................... 370
ЛИТЕРАТУРА ................................................................................................................. 372
Рекомендуемые учебники по дисциплине «Химия»
................................................................................... 372
Литературные источники информации
........................................................................................................... 372
Источники информации в системе Интернет:
............................................................................................... 375
Предисловие
Данное учебное пособие разработано на основе курса лекций по химии, которые читаются в Арсеньевском технологическом институте (филиале) ДВГТУ в течение нескольких лет. Курс лекций разработан на основе требований Государственных образовательных стандартов ХИМИЯ ЕН.Ф. 04 и ХИМИЯ ЕН.Ф. 05 для студентов соответствующих специальностей. В данном учебном пособии автор представляет учебный в строгом соответствии со структурой федерального компонента государственных образовательных стандартов по дисциплине «Химия» для студентов, обучающихся специальностям
151001 (Технология машиностроения), 140211 (Электроснабжение) и 080502 (Экономика и управление на предприятии (в машиностроении)), отказавшись от традиционного изложения предмета. При этом особое внимание уделялось тому, чтобы обеспечить системный подход (присущий духу государственного образовательного стандарта) и соответствие содержания и структуры данного учебного пособия тому объему знаний, который входит в предметную область и паспорт дисциплины.
Федеральный компонент: Требования Государственного образовательного стандарта к минимальному объему программы
Требования Государственного образовательного стандарта к минимальному объему программы дисциплины ХИМИЯ ЕН.Ф. 04
:
 Химические системы, растворы, дисперсные и электрохимические системы, катализаторы и каталитические системы; полимеры и олигомеры; химическая термодинамика и кинетика; энергетика химических процессов; химическое и фазовое равновесие; скорость реакции и методы ее регулирования; колебательные реакции; реакционная способность веществ; химия и периодическая система элементов; кислотные, основные и окислительно-восстановительные свойства веществ; химическая связь; комплиментарность; химическая идентификация; качественный и количественный анализ; аналитический сигнал; химический, физический и физико-химический анализ Химические системы, растворы, дисперсные и электрохимические системы, катализаторы и каталитические системы; полимеры и олигомеры; химическая термодинамика и кинетика; энергетика химических процессов; химическое и фазовое равновесие; скорость реакции и методы ее регулирования; колебательные реакции; реакционная способность веществ; химия и периодическая система элементов; кислотные, основные и окислительно-восстановительные свойства веществ; химическая связь; комплиментарность; химическая идентификация; качественный и количественный анализ; аналитический сигнал; химический, физический и физико-химический анализ
Химический практикум
По сравнению с Государственным стандартом ХИМИЯ ЕН.Ф. 04, д
ля студентов, обучающихся специальности «Экономика и управление на предприятии» Государственный образовательный стандарт ХИМИЯ ЕН.Ф. 05
дополнен следующими темами:
Основы биохимических процессов и их применение в технологической отрасли; теоретические основы мембранных технологий, современные мембранные материалы, перспективы развития мембранных технологий
Основная цель курса состоит в том, чтобы развить у студентов нехимических специальностей химическое мышление, не на профессионально-предметном, что, естественно, невозможно, а на концептуальном уровне, с тем, чтобы будущий специалист мог разобраться в сути химических проблем, возникающих в ходе его профессиональной деятельности.
Новизну данного учебного пособия можно усмотреть в авторской попытке внести в него все
компоненты государственных стандартов для указанных специальностей, включая такие темы, как колебательные реакции, химическая идентификация, основы биохимических процессов и теоретические основы мембранных технологий с тем, чтобы студенты могли найти весь базовый учебный материал в одном пособии.
Как известно, преподавание химии в нехимическом вузе отличается от школьного уровня не столько широтой охвата материала, сколько глубиной его рассмотрения, хотя эта глубина и несравнима с той, которая требуется при преподавании химии в химическом вузе. Особенностью данного учебного пособия является то, что автор стремился уделить особое внимание вопросам, позволяющим обобщить и объяснить материал по составу и свойствам вещества, с учетом междисциплинарных связей и задач отраслевой практики, сознательно сократив описательную часть курса. В данном пособии была сделана попытка сделать содержание пособия соответствующим самым современным представлениям об изучаемом предмете, а изложение учебного материала убедительным и доказательным для студентов, а также установить связь излагаемого материала с другими фундаментальными и смежными дисциплинами (такими, например, как физика и материаловедение).
Дисциплина «Химия» читается на первом курсе в первом семестре, поэтому необходимые требования к уровню освоения содержания дисциплины включают усвоение основ химии в объеме средней школы. В данном учебном пособии не предусмотрено применение элементов высшей математики, так как пособие предназначено для студентов первого курса, которые еще не владеют соответствующим математическим аппаратом.
Особое внимание в процессе разработке данного учебного пособия уделялось тому, чтобы сделать его доступным для понимания студентов, с тем, чтобы каждый студент в результате изучения предмета «Химия» в рамках данного курса:
Получил представление о современных взглядах на строение вещества в рамках системного подхода;
Разобрался в главных закономерностях химических процессов;
Научился ориентироваться в вопросах химии, имеющих наибольшее значение для его будущей специальности;
Овладел техникой несложного химического эксперимента посредством химического практикума (предусмотренного Государственным стандартом для студентов технических специальностей).
Для облегчения этой задачи теоретические положения в данном пособии иллюстрируются примерами, приводятся задания для самостоятельной работы, даются рекомендации по выполнению заданий для успешной самостоятельной работы студентов над предметом. Задания, предлагаемые в данном пособии, составлены с учѐтом требований, предъявляемым студентам при Интернеттестировании на федеральном уровне.
Объѐм химических знаний, представленных в данном учебном пособии, ограничен. Оно не может полностью заменить фундаментальный учебник по химии, однако его можно рассматривать именно как пособие, долженствующее облегчить знакомому со школьным курсом химии учащемуся, в том числе и самостоятельно изучающему предмет, успешное усвоение учебного материала фундаментальных учебников, рекомендованный список которых, вместе с перечнем дополнительной литературы, приводится в данном учебном пособии. Пособие также поможет студентам восполнить пробелы в школьном химическом образовании.
Введение
Химия как раздел естествознания
Химия как наука является разделом естествознания
- комплекса наук о природе, о мире в его естественном состоянии, независимо от человека. В этот комплекс входят физика и астрономия, химия и биология, геология и география, а также многочисленные приложения этих наук. Химия - это естественная наука, изучающая химические превращения материи, существующей в виде вещества. Материей
мы называем всѐ, что окружает нас (и нас самих), действует на наши органы чувств (или на приборы) и отражается нашим сознанием. По современным представлениям, вещество является только одной из трех форм материи; двумя другими формами являются поле и вакуум
. В современной физике веществом называют форму материи, состоящую из элементарных частиц, имеющих собственную массу (массу покоя
), а именно, из фермионов
, которые образуют электронные оболочки и атомные ядра, атомы и молекулы. При химических процессах происходит обмен атомами между различными веществами, перераспределение электронов, разрушение одних химических связей и возникновение других. Химические процессы являются проявлением электромагнитного взаимодействия
, переносчиком которого служит фотон.
При физическом изменении вещества не происходит изменений его внутреннего строения, состава и свойств. В результате химических изменений вещества (химической реакции) происходят изменения не только физического состояния веществ (например, его агрегатного состояния), но меняется также химический состав и структура веществ. С углублением знаний о строении материи эволюционировало и понятие «химическая реакция». По современным представлениям, химическая реакция – это процесс, в результате которого изменяются состав, структура или заряд участвующих в процессе частиц (атомов, молекул, ионов или радикалов) при неизменности химической природы атомов.
Химия изучает разнообразные химические процессы, и эти знания помогают человеку получать необходимые для его жизнедеятельности материалы и двигаться по пути материально-технического прогресса.
Значение химии в изучении природы принципиально важно, так как эта наука отвечает на вопрос о строении и свойствах веществ, из которых состоят все объекты природы, в том числе и сам человек. По словам А. Эйнштейна, только теория решает, что именно мы ухитряемся наблюдать
,
поэтому «нет ничего практичнее хорошей теории». Химия составляет теоретическую основу любого химического производства, от переработки природного сырья (металлических руд, нефти, природного газа) до изготовления бытовых товаров. Понимание законов химии и умение их использовать на практике необходимо каждому инженеру, технологу или экономисту, потому что нет ни одного производства, где не применялись бы те или иные химические процессы (получение чистых металлов и других материалов, нанесение покрытий, очистка воды и т.д.). Любая промышленно развитая страна имеет развитую промышленность, а тем самым обязана тратить большие средства на развитие теоретической химии.
Основные задачи современной химии
Цель любых научных исследований состоит в изучении законов природы для последующего их использования в практической деятельности. Практическое применение химической теории сводится к решению трех основных проблем:
1. Получения максимального количества вещества с заданными свойствами с минимальными затратами исходных веществ и энергии на осуществление процесса; 2. Получения максимального количества энергии (теплоты или электричества) для дальнейшего ее использования;
3. Осуществления всех химических процессов с оптимальной скоростью.
ЛЕКЦИЯ 1. Химические системы
1.1. Вещество как система
Применение теории систем к решению конкретных проблем называют системно-структурным подходом
. Этот подход применим и в химии.
Системой
вообще называется множество элементов, находящихся в таких отношениях друг с другом, которые придают множеству определенную целостность и единство, то есть взаимодействуют меж собой
.
Системным подходом
называют такой способ рассмотрения проблемы, при которой ее состояния, свойства и особенности выводятся из состояния и свойств составляющих ее элементов.
Системе свойственна целостность
, то есть свойство изменяться при изменении свойств ее элементов. Изменение любого элемента системы оказывает воздействие на все другие элементы системы и изменяет всю систему. И наоборот, воздействие на систему оказывает влияние на свойства составляющих ее элементов. При объединении элементов в целостную систему у них появляются новые свойства, то есть целое не является механической суммой частей. Примером может служить молекула воды, составленная из двух атомов водорода и одного атома кислорода. Свойства воды не есть линейная комбинация свойств водорода и кислорода, у воды появляются новые, присущие только ей характеристики. Такое свойство системы называется эмерджентностью
(от английского слова emerge
– внезапно возникать)
Любой объект окружающего нас мира представляет собой целое в одном отношении и часть целого в другом. Любую систему можно расчленить на элементы, которые, в свою очередь, могут рассматриваться как система. В то же время сама данная система может выступать в виде элемента другой более широкой системы. Это свойство систем называют их иерархическим строением
. Следствием иерархического строения является возможность изучения систем при последовательном переходе с одного уровня расположения элементов системы на другой. Самой большой и сложной является наш мир в целом, то есть система под названием Вселенная. Во всей этой системе в целом и в каждой ее части материя находится в непрекращающемся движении
. Мерой движения материи является энергия,
а также ее превращения из одной формы в другую.
Химия как наука о веществе должна, прежде всего, дать ответ на вопрос, как вещество устроено. Системный подход можно применить и к поискам ответов на этот вопрос. Если считать вещество сложной системой, на вопрос, из чего состоит данная сложная система, следует дать ответ – из более простых систем. Отсюда следует, что в данном случае логично применить системный подход с иерархическим усложнением, переходя от более простых систем к системам более сложным
. Однако при любом подходе необходима концепция
(что в переводе с латинского языка означает «понимание»), то есть основополагающее допущение. Таких концепций о строении вещества существует две, и обе они зародились ещѐ в глубокой древности, в V –
IV вв. до нашей эры в Древней Греции.
1.2. Концептуальный уровень
Одна из двух основополагающих идей о строении вещества называется корпускулярной концепцией
. Суть еѐ состоит в том, что любое вещество можно раздробить на первичные структурные элементы, то есть существует предел деления
материи. Автором корпускулярной концепции читается греческий философ Демокрит (460370 гг. до н.э), который утверждал, что на свете нет ничего, кроме атомов и пустоты
. Рассуждая о том, почему при сильном нагревании улетучивается вода, как можно ощутить запах цветка на расстоянии и куда девалась позолота с рук статуи Зевса в местном храме, Демокрит пришел к выводу о том, что тела только кажутся нам сплошными, а на самом деле состоят из мельчайших частиц – атомов. В переводе с греческого языка слово «атомос»
означает «неделимый»
.
Другой древнегреческий философ, Аристотель (384-323 гг. до н.э.), считал, что природа не терпит пустоты, и, отвергая пустоту, вместе с ней отрицал и существование атомов Демокрита. Аристотель считал, что четыре стихии (тепло, холод, сухость и влажность), соединяясь попарно, составляют четыре элемента мира: воду, воздух, огонь и землю
. Первоосновой же, или первоматерией мира, является идеальный пятый элемент - эфир. Эту концепцию называют континуальной концепцией
строения вещества (других форм материи древние не знали) от латинского слова «континуум», то есть нечто сплошное и неделимое (как эфир). Следовательно, из континуальной концепции строения вещества вытекает вывод, что предела
деления материи не существует
. Ещѐ одним выводом, из учения Аристотеля, который взяла на вооружение алхимия средневековья, была идея о возможности трансмутации,
то есть превращения одних элементов в другие, например, свинца или ртути в золото, или наоборот.
Какая же концепция строения верна? Решить это вопрос можно только опытным путѐм. Разлагая самые разные вещества на составные части, экспериментаторы убедились в том, что Аристотель неправ. Вода, например, не является простым элементом, так как из неѐ можно выделить ещѐ более простые субстанции – водород и кислород. Воздух также является сложной системой и состоит из смеси различных газов, основными компонентами которой являются азот и кислород. Земля состоит из множества веществ, как простых, так и сложных, некоторые из них по традиции даже называются «землями».
Континуальная концепция строения вещества зашла в тупик, и альтернативой ей послужил атомизм, о котором всѐ чаще вспоминали мыслители XVI-XVII веков. Так, например, в 1647 году Пьер Гассенди
(1592-1655), французский ученый и философ, не только отверг представления Аристотеля об устройстве материи в виде вещества, но и объяснил на основе учения древних атомистов
, каким именно образом возникают в мире многие миллионы разнообразных тел. Для этого, утверждал Гассенди, не требуется большого количества атомов. Атом - это все равно, что строительный материал для домов. Как из строительных материалов - кирпичей, досок гвоздей - можно построить и бедную хижину, и величественный дворец, так из нескольких десятков различных атомов природа может построить тысячи тысяч разнообразных тел. При этом различные атомы в каждом теле объединяются в небольшие группы, которые Гассенди назвал молекулами
, от латинского слова moleс
(«массочка»).
В 1668 году Роберт Бойль(1627-1691), назвал истинными элементами, в отличие от алхимических «элементов мира», «... некоторые первоначальные и простые, вполне не смешанные тела; эти тела не состоят из других тел или друг из друга и являются составными частями, из которых сложены все вполне смешанные тела и на которые последние, в конце концов, распадаются»,
то есть определил предел качественного деления вещества, элемент
. Количественным же пределом деления материи считались атомы
.
Приверженцем атомного строения материи был Исаак Ньютон (1643 1727). «Мне представляется
, - писал он, - что Бог с самого начала сотворил вещество в виде твердых, непроницаемых, подвижных частиц и что этим частицам он придал такие размеры и такую форму, и такие другие свойства, и создал их в таких относительных количествах, как Ему нужно было для той цели, для которой он их сотворил…»
. Огромный вклад в развитие атомно-молекулярного учения внѐс Михаил Васильевич Ломоносов (1711 - 1765). Ломоносов считал, что все тела состоят из атомов (правда, атомы он ошибочно называл элементами) и их сочетаний - молекул, которые он называл корпускулами. Ломоносов, одновременно с французским химиком Лавуазье, доказал закон сохранения вещества и позднее обобщил его до закона сохранения материи и движения. Природу теплоты Ломоносов объяснял вращательным и колебательным движением атомов и молекул. Следующий шаг в познании строения материи сделал английский химик, физик и метеоролог Джон Дальтон (1766 - 1844). Дальтон считал, что атомы различных элементов имеют различную массу
. Изучая растворимость газов в жидкости, он вычислил относительные веса таких атомов, как азот, углерод, кислород и сера, взяв за единицу вес атома водорода. Он также вычислил молекулярные массы некоторых сложных веществ. Существование атомов и молекул стало научным фактом, после того, как Дальтон их «взвесил», а итальянский физик и химик Амедео Авогадро (1776 -1856) «сосчитал». По закону Авогадро, в количестве любого вещества, равном его молекулярной или атомной массе (это количество вещества называют молем
) содержится одинаковое число атомов
или молекул
, а именно, число Авогадро 6,02∙10 23
. Один моль любого газа занимает объѐм 22,4 литра – именно благодаря этому обстоятельству и удалось «сосчитать» атомы и молекулы. «Увидеть» молекулы удалось в 1827 году английскому ботанику, почѐтному члену Петербургской Академии наук, Роберту Броуну (1773 -1858). Броун наблюдал в микроскоп взвесь очень мелких частиц пыльцы растений в воде и установил, что они находятся в непрерывном хаотическом движении. Причиной этого движения является непрерывная «бомбардировка» видимых в микроскоп частиц пыльцы невидимыми из-за своих малых размеров молекулами воды. Правда, механизм броуновского движения, вызванного вечным движением молекул, Альберт Эйнштейн объяснил только в 1905 году.
Ещѐ не так давно считалось, что увидеть атомы невозможно. Однако с изобретением в конце 20 века сканирующих микроскопов со сверхвысоким разрешением такая возможность появилась, и наше поколение отличается от предыдущих поколений, прежде всего тем, что мы увидели атомы, из которых состоим (Рис. 1.1)
Рис. 1. 1. Кристалл кремния при увеличении в 100 миллионов раз
 Фотография кристалла кремния сделана посредством сканирующего электронного микроскопа особого типа. Отдельные атомы (большие красно-жѐлтые треугольники) на поверхности кристалла расположены в строгом порядке, соединѐнные химическими связями (белые «облака»). Атомы не имеют строго определенных границ вследствие волновой природы электронов. Фотография кристалла кремния сделана посредством сканирующего электронного микроскопа особого типа. Отдельные атомы (большие красно-жѐлтые треугольники) на поверхности кристалла расположены в строгом порядке, соединѐнные химическими связями (белые «облака»). Атомы не имеют строго определенных границ вследствие волновой природы электронов.
К середине XIX, когда было открыто 74 элемента с различными свойствами и появилось стройное атомно-молекулярное учение, победу корпускулярной концепции над концепцией континуальной можно было считать окончательной.
Суть атомно-молекулярного учения можно выразить следующим образом:
Существует предел деления материи – атом;
Атомы отличаются друг от друга массой и размерами, но внутренней структуры не имеют (неделимы);
Атомы с одинаковой массой образуют химический элемент;
Атомы соединяются друг с другом в соответствии со своей валентностью и образуют молекулы;
Валентностью называют свойство атомов одного элемента соединяться с определѐнным числом атомов другого
элемента;
Атомы и молекулы находятся в постоянном движении;
В совокупности, атомы или молекулы, состоящие из одинаковых атомов, образуют простые вещества. Эти вещества отличаются друг от друга по химическим свойствам (металлы и неметаллы);
В совокупности, молекулы, состоящие из разных атомов, образуют сложные вещества. Эти вещества отличаются по химическим свойствам (оксиды, кислоты, основания, соли и т.д.);
Процесс превращения одних молекул в другие в соответствующих условиях есть химическая реакция;
Превращение одних атомов в другие (трансмутация) невозможно.
В рамках корпускулярной концепции в виде атомно-молекулярного учения ответ на вопрос о строении материи при системном подходе с иерархическим усложнением можно представить следующим образом: Атом → Молекула → Вещество
В 1969 году Д.И. Менделеев (1834 -1907) открыл закон, объясняющий, как изменяются свойства химических элементов
, который является вершиной атомно-молекулярного учения. Попытки систематизации химических элементов предпринимались и до Менделеева, но они основывались на объединении элементов в группы только на основании их физического сходства и химических свойств.
Так, немецкий химик Дѐберейнер еще в 1829 г. заметил сходство между некоторыми элементами (хлором, бромом и йодом; литием, натрием и калием) и объединил их в триады. В 1964 английский химик Ньюлендс предложил «закон октав», по которому элементы объединялись в группы, причем все восьмые элементы в этих группах имели одинаковые свойства. Сходную таблицу предложил и немецкий химик и физик Лотар-Мейер.
Менделеев пришел к выводу, что в основу систематики элементов должна быть положена их относительная атомная масса
(атомный вес).
Массы атомов, выраженные в граммах, очень малы (атом
водорода, например, весит около 1,67∙10-24
г), и пользоваться этими величинами крайне неудобно. Поэтому еще в 1803 году английский химик Д. Дальтон предложил использовать соотношение весов, или относительную массу элементов, взяв за единицу массу самого легкого атома, водорода (водородную единицу). Впоследствии водородная единица была заменена кислородной (1/16 часть массы атома кислорода). В 1961 году за атомную единицу массы была принята 1/12 часть массы атома углерода (вернее, изотопа углерода 12
С). Атомные массы кислорода и водорода, например, равны соответственно 15,9994 и 1,00794. Размеры атомов оценивают в ангстрѐмах. Один ангстрѐм равен 10-10
м (10-8
см). Мы не можем видеть атомов, потому что они несоизмеримы с объектами «человеческих» размеров. Поэтому мир человека принято называть макромиром
, а мир атомов – микромиром
. Поскольку на микроуровне вещество (в рамках атомно-молекулярного учения) состоит из атомов, то его можно назвать микровеществом
. Расположив все известные в то время элементы (74) в порядке возрастания их атомных масс (весов), Менделеев обнаружил, что сходные в химическом отношении элементы встречаются через правильные интервалы (периоды), то есть химические свойства элементов периодически повторяются. Периодический закон Менделеев сформулировал следующим образом:
Свойства простых тел, а также формы и свойства соединений элементов, находятся в периодической зависимости от величины атомных весов элементов
.
Если распределить элементы в порядке возрастания их атомных масс по строкам (периодам) таким образом, чтобы в первой строке было 2 элемента, во второй и третьей – по 8, в четвѐртой и пятой – по 18, а в шестой и седьмой – по 32, то свойства элементов будут периодически повторяться. Каждый период начинается с очень активного металла, затем металлические свойства ослабевают, а неметаллические усиливаются, а заканчивается каждый период инертным газом. Это значит, что свойства элементов, расположенных в системе Менделеева определенным образом повторяются. Понятие о валентности
как о свойстве атомов соединяться в определѐнных соотношениях
, объясняло, например, строение молекулы воды Н2
О тем, что одновалентный водород соединяется с двухвалентным кислородом в соотношении 2 атома водорода к 1 атому кислорода.
Свойства элементов, расположенных в системе Менделеева определенным образом, повторяются, причѐм периодичность этих повторений не простая, а сложная и выражается числами 2, 8, 18 и 32. Менделеев не мог объяснить, откуда берутся эти «магические» числа.
Ответ на этот вопрос дала квантовая химия.
1.3. Субатомный уровень
Современная химия отвечает на вопрос о строении вещества в рамках корпускулярной концепции. Однако пределом деления вещества считаются не атомы, а дискретные
(от английского слова discrete
– разбивать) элементарные частицы
. То, что у атома есть составные части, удалось обнаружить, изучая электрические явления. В 1897 году английский физик Джозеф Джон Томсон, изучая природу катодных лучей, или электрического тока, открыл отрицательно первую элементарную частицу – электрон
, и, тем самым, доказал, что атом имеет структуру, то есть является делимым
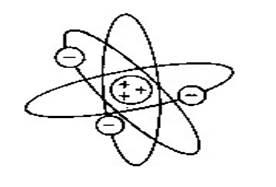
. Ученик Томсона, Эрнест Резерфорд (1871 -1937) в 1910 году открыл атомное ядро и предложил другую модель атома: планетарную
(см. Рис.1.2). В таком атоме, размеры которого составляют 10-10
м, вокруг небольшого по размеру (1015
-10-14
м), но очень массивного положительно заряженного ядра движутся отрицательно заряженные электроны.
Рис.1.2 Планетарная модель атома
Ядро, как выяснилось впоследствии, состоит из тяжѐлых элементарных частиц, протонов и нейтронов
. На сегодня физикам известно более 400 элементарных частиц, которые делятся на фермионы
и бозоны
. Фермионы считаются частицами, образующими вещество, тогда как бозоны являются частицами полей
 (электромагнитного, сильного и слабого ядерного и гравитационного) и переносчиками соответствующих фундаментальных взаимодействий. Все элементарные частицы можно классифицировать по массе, заряду, времени жизни и спину
. Для химии наиболее важными являются четыре элементарные частицы: это протон, нейтрон и электрон (фермионы), а также фотон
(бозон), который является переносчиком электромагнитного взаимодействия, так как все химические превращения имеют электромагнитную природу. Масса электрона me
оставляет 9,109410-28
г, масса протона равна 1,610-24
г, или 1836 me
) а масса нейтрона - 1840 me
. В атомных единицах масса протона и нейтрона равна 1, а масса электрона не принимается в расчѐт. Фотон не имеет массы покоя, так как нет такой системы отсчѐта, относительно которой можно было бы представить его покоящимся, однако в движении его масса отлична от нуля. Заряд протона равен +1, электрона -1, нейтрона - 0, для фотона понятия заряда не существует. Электроны, протоны и фотоны являются стабильными частицами, время жизни которых превышает 1027
лет. Свободный нейтрон является квазистабильной частицей, время его жизни составляет 1000 секунд. Все элементарные частицы с их невероятно малыми массами и размерами - это объекты микромира
, и их свойства не имеют аналогов в привычном для нас макромире
. Одним из таких свойств является спин (от английского слова spin
– веретено или волчок). Фермионы обладают полуцелым спином, так, например, спин электрона принимается равным ±1/2. У фотона, поскольку он является бозоном, спин целый и равен 1. Спин определяет собственный момент количества движения (электромагнитного, сильного и слабого ядерного и гравитационного) и переносчиками соответствующих фундаментальных взаимодействий. Все элементарные частицы можно классифицировать по массе, заряду, времени жизни и спину
. Для химии наиболее важными являются четыре элементарные частицы: это протон, нейтрон и электрон (фермионы), а также фотон
(бозон), который является переносчиком электромагнитного взаимодействия, так как все химические превращения имеют электромагнитную природу. Масса электрона me
оставляет 9,109410-28
г, масса протона равна 1,610-24
г, или 1836 me
) а масса нейтрона - 1840 me
. В атомных единицах масса протона и нейтрона равна 1, а масса электрона не принимается в расчѐт. Фотон не имеет массы покоя, так как нет такой системы отсчѐта, относительно которой можно было бы представить его покоящимся, однако в движении его масса отлична от нуля. Заряд протона равен +1, электрона -1, нейтрона - 0, для фотона понятия заряда не существует. Электроны, протоны и фотоны являются стабильными частицами, время жизни которых превышает 1027
лет. Свободный нейтрон является квазистабильной частицей, время его жизни составляет 1000 секунд. Все элементарные частицы с их невероятно малыми массами и размерами - это объекты микромира
, и их свойства не имеют аналогов в привычном для нас макромире
. Одним из таких свойств является спин (от английского слова spin
– веретено или волчок). Фермионы обладают полуцелым спином, так, например, спин электрона принимается равным ±1/2. У фотона, поскольку он является бозоном, спин целый и равен 1. Спин определяет собственный момент количества движения
элементарной частицы, не связанный с ее перемещениями как целого.
Иногда для объяснения понятия спина используют такую аналогию – электрон представляют как летящий волчок, или круговой ток, создающий собственное магнитное поле. Такая аналогия позволяет объяснить наличие спина  1/2 у электрона и протона, но не у нейтрона – частицы с нулевым зарядом. При всех способах его регистрации спин всегда направлен вдоль той оси, которую наблюдатель выбрал за исходную. Значение спина 1/2 означает, что электрон (протон, нейтрон) становится идентичным сам себе при обороте на 7200
, а не 3600
, как в нашем трехмерном мире. В результате «двойного поворота» создаваемое электроном магнитное поле вдвое больше того, которое мог бы дать вращающийся заряженный шарик. Спин принято считать одним из фундаментальных свойств материи, то есть он не является следствием других свойств природного объекта, также как, например, гравитация и электричество. 1/2 у электрона и протона, но не у нейтрона – частицы с нулевым зарядом. При всех способах его регистрации спин всегда направлен вдоль той оси, которую наблюдатель выбрал за исходную. Значение спина 1/2 означает, что электрон (протон, нейтрон) становится идентичным сам себе при обороте на 7200
, а не 3600
, как в нашем трехмерном мире. В результате «двойного поворота» создаваемое электроном магнитное поле вдвое больше того, которое мог бы дать вращающийся заряженный шарик. Спин принято считать одним из фундаментальных свойств материи, то есть он не является следствием других свойств природного объекта, также как, например, гравитация и электричество.
В рамках корпускулярной концепции на субатомном уровне (приставка «суб» означает, что речь идѐт об уровне, лежащем в глубинах атома) ответ на вопрос о строении материи при системном подходе с иерархическим усложнением можно представить следующим образом:
Элементарные частицы → ядра атомов + электроны, вращающиеся вокруг ядра = атомы → молекулы → вещество
В соответствии с ядерно-планетарной моделью атома Резерфорда, атом состоит из положительно заряженного ядра и электронов, количество которых равно заряду ядра, так что атом в целом является электронейтральным. Атомное ядро
– это система, состоящая из массивных частиц нуклонов
(от латинского слова «нуклеус» – «ядро»),
положительно заряженных протонов
(открыты в 1919 г. Э. Резерфордом) и не имеющих заряда нейтронов
(открыты в 1932 Дж. Чедвиком). Оказалось, что заряд ядра численно равен порядковому номеру атома в периодической системе по закону Мозли
, или закону атомного номера
. В 1913 г. английский ученый Генри Мозли установил: корень квадратный из частоты рентгеновского спектра элемента является линейной функцией порядкового номера элемента в периодической системе:
 c c
, (1.1)
где ν – частота рентгеновского излучения атома; λ – длина волны этого
излучения; с – скорость света (3∙108
м/сек); Z - порядковый номер элемента, К и α – константы серий линий спектра.
Это означает, что, зная порядковый номер элемента в системе Менделеева (например, элемента №5), можно установить, как устроен атом данного элемента. Заряд ядра этого атома, в соответствии с законом Мозли, равен +5, следовательно, в ядре атома элемента №5 находится пять протонов, а вокруг ядра вращается пять электронов. Число нейтронов в ядре может меняться (для лѐгких элементов оно чаще всего равно числу протонов). Это открытие устраняло кажущееся противоречие в системе Менделеева, где некоторые элементы с большей массой стояли впереди элементов с меньшей массой (теллур и йод, аргон и калий, кобальт и никель). Оказалось, что противоречия здесь нет, так как заряды ядер этих элементов точно соответствуют их положению в периодической системе. Заряд атомного ядра
оказался той основной величиной, от которой зависят свойства химического элемента. Поэтому периодический закон Менделеева в современной формулировке звучит так:
Свойства элементов и образуемых ими простых и сложных веществ находятся в периодической зависимости от заряда ядра атомов элементов
.
Химическим элементом
по современным представлениям считается совокупность атомов с одинаковым числом протонов в ядре, или с одинаковым зарядом ядра
.
Прибавляя к ядру протоны или выбивая их из ядра (например, в ускорителях элементарных частиц), можно из одного элемента получить другой, то есть осуществить ту трансмутацию
, о которой мечтали алхимики. Именно поэтому создатель науки об атомном ядре Э. Резерфорд назвал свою последнюю книгу «Современной алхимией». Однако для такой трансмутации нужна колоссальная энергия, которую нельзя получить в какой бы то ни было химической реакции, поэтому такие ядерные реакции
осуществляются в ускорителях элементарных частиц. При этом теоретически можно получить сколь угодно много элементов, однако для химии такие элементы, во всяком случае, на данном этапе, никакого практического значения не имеют, так как их атомы получают в ускорителях элементарных частиц буквально поштучно, а время жизни таких атомов ничтожно мало. Ядерные реакции в данном учебном пособии не рассматриваются.
Сумма масс протонов и нейтронов составляет массовое число
, или атомную массу А
элемента. Ядра, имеющие одинаковое число протонов, но различное число нейтронов, называются изотопами.
Химические свойства изотопов одинаковы: это один и тот же химический элемент. Любой элемент состоит из совокупности изотопов: например, водород – это смесь изотопов с массовыми числами 1 (протий), 2 (дейтерий) и 3 (тритий), поэтому приведенная в системе Менделеева атомная масса элемента представляет собой не целое, а дробное число (для водорода – 1,008). Многие изотопы неустойчивы и распадаются с выделением излучения или частиц с высокой энергией; это явление называется естественной радиоактивностью
.
Встречаются также ядра, массовые числа которых одинаковы, но число протонов в ядре разное. Такие ядра называются изобарами
; изобары обладают различными химическими свойствами, следовательно, являются различными элементами.
В химических превращениях ядро, спрятанное в глубинах атома, непосредственного участия не принимает. Поэтому химику важнее всего знать, как располагаются электроны в том или ином атоме, так как именно они обуславливают химические превращения элементов. Количество электронов
на атомных орбитах равно количеству протонов
в ядре, или заряду ядра, так как протон заряжен положительно (его заряд принимается равным +1), электрон – отрицательно (-1), а атом в целом электронейтрален. Так, у атома №5 вокруг ядра вращаются пять электронов.
1.4. Квантовый уровень
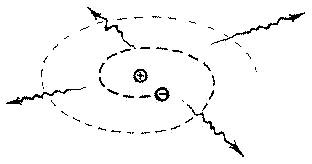
Планетарная модель атома противоречит законам классической электродинамики, то есть теории движения заряженных тел. В соответствии с этими законами, движущийся по орбите электрон очень скоро (через 10-8
сек) должен упасть на ядро (см. Рис. 1.3).
Рис. 1. 3. Вращаясь по замкнутой орбите, электрон должен постоянно излучать энергию
Однако на самом деле атомы очень стабильны. Датский физик Нильс Бор (1885-1962) разрешил это противоречие, предложив свою модель атома, основанную на двух постулатах, которые называют постулатами Бора
:
атом имеет прерывистую последовательность стационарных состояний, находясь на которых электрон не испускает и поглощает энергию;
при переходах от состояния к состоянию электрон излучает квант электромагнитной энергии.
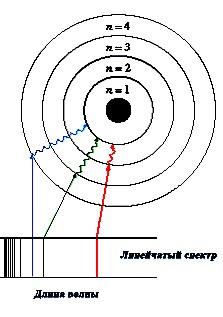
На основании своей модели атома водорода Бор сумел объяснить
линейчатые спектры атомов. Если атомы какого-либо элемента, например, водорода, возбудить (сильно нагреть), то они начинают испускать свет. Этот свет можно разложить в спектр (при помощи стеклянной призмы или дифракционной решетки) и получить ряд линий (цветных, если они расположены в видимой области спектра) – «визитную карточку» атома (см. Рис. 1.4). Каждый элемент имеет уникальный спектр, что широко используется для определения элементного состава различных образцов, а сыграло огромную роль в открытии новых элементов. Так, например, при анализе спектра Солнца был открыт гелий. Такой метод исследования элементов и их соединений называется спектральным анализом
. Твердые тела и жидкости, где атомы тесно сближены друг с другом, дают сплошной спектр, а газы и пары, где атомы удалены друг от друга, - линейчатый, содержащий только определенные длины волн. Спектры и спектральный анализ были открыты ещѐ в XVIII веке и широко применялись в научных исследованиях, однако только Бору удалось объяснить происхождение и уникальность линейчатых спектров атомов.
Рис 1.4. Спектр водорода в видимой области

Наиболее простой спектр имеет атом водорода, поэтому он очень хорошо изучен. В видимой области спектра имеются всего четыре линии, - красная, зеленая, синяя и фиолетовая (Рис.1.4).
Бор предположил, что каждая спектральная линия появляется в результате перехода электрона в атоме водорода с более удалѐнной орбиты на орбиту, более близкую к ядру. При этом испускается порция энергии
, равная разности энергий электрона на более удалѐнной и менее удалѐнной орбитах (Рис. 1.5).
Рис 1.5. Модель Бора, объясняющая появление в спектре водорода строго определенных линий
 Теоретические расчѐты частот спектральных линий водорода, сделанные Бором, точно совпали с экспериментально полученными значениями. Это значит, что модель Бора отражает реальность. Энергия каждого перехода рассчитывается по формуле Планка-Эйнштейна: Теоретические расчѐты частот спектральных линий водорода, сделанные Бором, точно совпали с экспериментально полученными значениями. Это значит, что модель Бора отражает реальность. Энергия каждого перехода рассчитывается по формуле Планка-Эйнштейна:
E
 (1.2) (1.2)
Где Е
– энергия, v –
частота спектральной линии, а h
(постоянная Планка), или квант действия
.
Понятие «действие» является одним из главнейших понятий механики. На современном языке оно звучит так: ничто на свете не происходит без затрат энергии и времени, но особенно важно их произведение. Оно показывает, что малая энергия за долгое время производит то же действие, что большая энергия за короткий срок.
Произведение энергии и времени есть действие
. В восемнадцатом веке Пьер-Луи де Мопертюи сформулировал классический принцип наименьшего действия «Если в природе происходит само по себе какое-либо изменение, то необходимое для этого количество действия есть наименьшее возможное»
, который спустя столетие стал руководящим в классической механике; из него выводились все уравнения движения. Мерой движения тел является энергия. Энергия существует в разных формах, например, в виде тепла и света. О природе света спорили еще Ньютон и его современник Гюйгенс. Ньютон считал свет состоящим из материальных частиц
, как все остальные материальные тела. Гюйгенс же рассматривал свет как волновой процесс. В XIX было доказано, что свет - это электромагнитные волны
с определенной длиной волны λ
и частотой ν
. Ниже в Таблице 1.1 приведен весь диапазон электромагнитных волн. По мере продвижения к рентгеновским лучам дина волны уменьшается, а частота растѐт. Область видимого света по сравнению со всем диапазоном электромагнитных волн очень мала.
Таблица 1.1. Диапазон электромагнитных волн
| Вид излучения
|
Длина волны
|
Частота
|
| Постоянный ток
|
3х106
м
|
102
гц
|
| Переменный ток
|
3х104
м
|
104
гц
|
| Радиоволны
|
3х102
- 3 м
|
106
-108
|
| Инфракрасное излучение
|
3х10-4
|
1012
|
| Видимый свет:
Красный
Синий
|
3х10-6
3х10-8
|
10
14
1016
|
| Ультрафиолетовое излучение
|
3х10-9
|
1017
|
| Рентгеновское излучение
|
3х10
-10
|
1018
|
| Гамма-излучение
|
3х10
-12
|
1020
|
В 1900 году немецкий физик Макс Планк (1858 - 1947) высказал предположение, что вся энергия, в том числе и тепловая, и световая, излучается и поглощается в виде порций, или квантов
(в основе этого термина лежит латинское слово «quantum», что значит «сколько»). Наименьшую порцию энергии, равную 6,6262∙10-34
Дж∙сек
, называют квантом действия
, или постоянной Планка. В 1905 году Альберт Эйнштейн (1879 - 1955) пришѐл к выводу, что свет
существует в виде потока квантов, или фотонов
, каждый из которых является одновременно волной и частицей, то есть обладает корпускулярноволновым дуализмом (
двойственностью)
.
Только с помощью такого допущения он смог объяснить механизм фотоэффекта: облученные светом определенной частоты, некоторые вещества начинали испускать электроны, то есть проводить электрический ток, потому, что только кванты света, обладающие достаточной энергией, способны «выбить» из вещества электроны.
Энергию фотона можно вычислить по формуле Планка-Эйнштейна (1.2). Отсюда можно вывести физический смысл мировой квантовой константы h
. Чем выше частота излучения, тем большей энергией обладает квант. У каждого кванта частота колебаний своя, но основа дробности и малости у всех квантов общая. Ею служит величина наименьшего физического действия в природе
. Для обозначения этого наименьшего действия Планк и ввел свою константу h
. Наука о движении в микромире получила название квантовой механики
, потому что постоянная Планка входит во все еѐ основные принципы и уравнения.
Оказалось, что электрон, как элемент микромира
, не подчиняется законам классической механики. Он не излучает, находясь на стационарной боровской орбите, а между боровскими орбитами находятся запрещѐнные области пространства, в которых электрон существовать не может. Неклассическая теория движения, квантовая механика
, составной частью которой является неклассическая теория строения вещества, квантовая химия
, объясняет это невероятными для классической физики свойствами электрона: он является одновременно и частицей, и волной
, то есть микрочастицам так же, как и фотонам света, присущ корпускулярно-волновой дуализм
. Электрон может находиться только в тех областях пространства, которые вмещают целое число электронов-волн. Длину волны электрона можно рассчитать по уравнению, предложенному французским физиком Луи де Бройлем (1892 - 1986):
 h
, (1.3) h
, (1.3)
mv
где h – постоянная Планка, равная 6, 6262∙10-34
Дж∙сек; m – масса
электрона, равная 9, 108∙10-34
г., а v – скорость движения электрона.
В соответствии с принципом неопределенности, который был сформулирован немецким физиков Вернером Гейзенбергом, для электрона (а в общем случае для любой микрочастицы) невозможно одновременно точно определить координату частицы Х и скорость V, или импульс Рх .
Математически этот принцип записывается так:  h
,
(1.4) h
,
(1.4)
 где х
и vх
– неопределенность[1]
координаты и импульса (скорости где х
и vх
– неопределенность[1]
координаты и импульса (скорости
движения).
Это значит, что невозможно
точно определить положение отдельного электрона в атоме, можно только предсказать с некоторой вероятностью положение его на определенной орбите
. В микромире действуют не динамические
, а вероятностные
законы.
Почти одновременно с Гейзенбергом Н. Бор сформулировал свой принцип дополнительности, в котором идея корпускулярно-волнового дуализма выражена в общем виде: «Понятие частицы и волны дополняют друг друга и в то же время противоречат друг другу, они являются дополняющими картинами происходящего».
Это значит, что микрочастица не может проявлять свойства частицы и волны одновременно, то есть корпускулярная и волновая картины должны дополнять друг друга, то есть быть комплиментарными
. Только при учете этих двух аспектов можно создать общую картину микромира.
Математически положение электрона-волны в атоме описывается уравнением Шрѐдингера
, или так называемой пси-функцией. В простейшем случае это волновое уравнение имеет вид:
 0,
(1.5) 0,
(1.5)
 где U - потенциальная энергия частицы; E – еѐ полная энергия; X, Y, Z - координаты частицы; - пси-функция; h - постоянная Планка где U - потенциальная энергия частицы; E – еѐ полная энергия; X, Y, Z - координаты частицы; - пси-функция; h - постоянная Планка
Квадрат модуля пси-функции  определяет вероятность обнаружения электрона в данной точке пространства в определенное время
. Решая уравнение Шрѐдингера (решение его для атомов сложнее водорода является очень трудной математической задачей), находят главное квантовое число
n, характеризующее те орбиты, нахождение на которых электронов любого атома периодической системы наиболее вероятно. определяет вероятность обнаружения электрона в данной точке пространства в определенное время
. Решая уравнение Шрѐдингера (решение его для атомов сложнее водорода является очень трудной математической задачей), находят главное квантовое число
n, характеризующее те орбиты, нахождение на которых электронов любого атома периодической системы наиболее вероятно.
Главное квантовое число n
определяет энергию электрона на той или иной орбите, или энергетическом уровне
. Значение n для электронов в каком-либо атоме можно найти, определив квадрат модуля пси-функции. Вычислить энергию электрона на определенном энергетическом уровне можно по следующему соотношению, которое вытекает из квантовой теории:
Z
2
 E
13,60 2
, (1.6) E
13,60 2
, (1.6)
n
где Z – заряд ядра атома; n – номер энергетического уровня.
Знак « - » показывает, что отрыв электрона требует затраты энергии. Энергия выражается в электрон-вольтах
. Один электрон-вольт – это энергия, необходимая для отрыва от атома первого электрона. Энергия электрона, главным образом, зависит от расстояния его до ядра (чем ближе к ядру находится электрон, тем меньше его энергия). Поэтому говорят, что главное квантовое число определяет положение электрона на том или ином энергетическом уровне
(или квантовом слое). Эти уровни обозначаются цифрами или буквами:
Значение главного квантового числа n: 1 2 3 4 5 6 7
Обозначение уровня: K L M N O P Q
В квантовой механике электрон представляют в виде электронного облака
определенной формы. Форма облака определяется орбитальным, или азимутальным квантовым числом l
, которое принимает значения от 0 до n-1, всего n значений и обозначается так:
Значение l
: 0 1 2 3
Обозначение: s p d f
Исторически первые четыре значения имеют буквенные символы, произошедшие от спектроскопических терминов, использованных в 1890-е годы при описании спектров щелочных металлов: 0 – s (sharp – резкий); 1 – p (principal – главный); 2 – d (diffuse – диффузный); 3 – f (fundamental – фундаментальный). Эти буквы не являются сокращениями слов, описывающих «форму» орбитали.
При l = 0 s - электронное облако имеет шаровую симметрию, при l = 1 pэлектронное облако имеет форму гантели, при l = 2 d – электронное облако можно представить в виде двух гантелей, ориентированных по двум взаимно перпендикулярным осям Х и У, при l = 3 f- облако представляют в виде двух гантелей, ориентированных по трѐм взаимно перпендикулярным осям Х, У и Z. Более сложных электронных облаков не существует.
Для того чтобы определить квантовое число, необходимо перебрать все значения главного квантового числа n
от 0 до (n-1). Это означает, что на первом, ближайшем к ядру, энергетическом уровне могут существовать только s-электроны, на втором – s- и p- электроны, на третьем – s-, p- и d- электроны, а начиная с четвѐртого – s-, p-, d- и f- электроны.
Магнитное квантовое число ml
определяет ориентацию электронного облака в пространстве. Магнитное квантовое число связано с орбитальным квантовым числом и для его определения нужно перебрать все значения орбитального числа l
+l
до -l
, включая 0. Таким образом, число его возможных значений равно 2l
+1. Для s-электронов при l
=0 существует только одно значение магнитного квантового числа (ml
= 0). Для р- электронов (l
=1) существует уже три значение магнитного квантового числа (+1, 0, -1). Это указывает на то, что рэлектроны отличаются по ориентации их в пространстве (то есть по энергии) по осям x, y и z, то есть энергетический уровень р-электронов расщепляется на три подуровня
(такое расщепление наблюдается при воздействии магнитного поля и называется эффектом Зеемана
). У d- и f-электронов существует соответственно 5 и 7 подуровней. Электрон также обладает свойством, которое называется спином
(от английского слова «spin», что значит «вращение» или «веретено»). Очень упрощенно спин можно считать вращением электронного облака вокруг своей оси по часовой стрелке или в противоположном направлении. Спин определяется спиновым квантовым числом ms
, которое может принимать значения +1/2 и -1/2. Положительное и отрицательное значение спинового кантового числа определяют противоположные направления спина. Электроны, имеющие одинаковое направление спина, называются параллельными (с параллельным спином), а противоположное направление спина - антипараллельными (с антипараллельным спином).
Набор квантовых чисел определяет положение любого электрона в любом атоме. Атомы расселяются по орбитам в соответствии с законами квантовой механики. При этом необходимо применять принцип Паули
, сформулированный в 1925 году швейцарским физиком Вольфгангом Паули: В атоме не может быть двух электронов, у которых были бы одинаковы все четыре квантовых числа.
Например, электроны с квантовым числом n=1 могут отличаться только спиновым квантовым числом (при n=1 l=0 и ms
=0). Понятно, что на этом уровне может располагаться только два s-электрона. Электронную конфигурацию этого уровня условно записывают в виде 1s2
.
Согласно правилу Гунда
, в подуровне (p, d или f)электроны стремятся занять энергетические состояния таким образом, чтобы суммарный спин был максимальным.
Согласно правилу Клечковского
, или принципу наименьшей энергии, заполнение энергетических уровней происходит в порядке возрастания суммы квантовых чисел n+l, а при их равенстве в порядке возрастания n, или в следующем порядке:
1s<2s<2p<3s<3p<4s<3d<4p<5s<4d<5p<6s<5d1
<4f<5d<6p<7s<6d2
<5f<6d<7p
Нарушение порядка заполнения орбиталей связано с тем, что энергия электронов зависит не только от заряда ядра, но и от взаимодействия между электронами, их взаимного отталкивания, или экранирования. У многоэлектронных атомов иногда наблюдаются исключения из правила Клечковского.
Набор квантовых чисел, принцип Паули, правила Гунда и Клечковского позволяют определить расположение электронов в любом атоме любого элемента в системе Менделеева. Такая «квантовая визитная карточка» атома определяет все его химические (и многие физические) свойства, поэтому очень важно научиться быстро и правильно «расписывать» по уровням (или «квантовать») электроны атома любого элемента.
Совокупность положений электрона в атоме, которая определяется набором значений квантовых чисел, называют атомной орбиталью (АО).
Для микрочастиц, к числу которых относятся электроны, нельзя точно определить их положение в определенный момент времени, но можно решить уравнение Шрѐдингера и определить вероятность нахождения электрона на том или ином расстоянии от ядра, то есть на той или иной атомной орбитали (или в квантовой ячейке
).
Рассмотрим атом водорода, в котором имеется всего один электрон. Решив для него уравнение Шрѐдингера[2]
(а решением является нахождение значение квадрата модуля пси-функции, в данном случае это  1), можно показать, что вероятность найти электрон на близком расстоянии от ядра ничтожно мала (близок к нулю квадрат модуля пси-функции, или плотность вероятности). Ничтожно мала и вероятность найти электрон на большом расстоянии от ядра, но на некотором расстоянии от него вероятность найти электрон максимальна. Расстояние это для атома водорода равно 0,053 нм. Это значение и считается радиусом ближайшей к ядру орбиты электрона. Так как 1, то главное квантовое число n единственного электрона атома водорода равно 1. Следовательно, орбитальное число равно 0, что соответствует шарообразной форме электронного облака, то есть единственный электрон атома водорода является s-электроном (размеры и форма электронного облака определяются распределением квадрата модуля пси-функции). S-орбиталь не расщепляется на подуровни, так как магнитное квантовое число при этом имеет одно значение ml
= 0. Спиновое квантовое число может принимать любое значение – либо 1), можно показать, что вероятность найти электрон на близком расстоянии от ядра ничтожно мала (близок к нулю квадрат модуля пси-функции, или плотность вероятности). Ничтожно мала и вероятность найти электрон на большом расстоянии от ядра, но на некотором расстоянии от него вероятность найти электрон максимальна. Расстояние это для атома водорода равно 0,053 нм. Это значение и считается радиусом ближайшей к ядру орбиты электрона. Так как 1, то главное квантовое число n единственного электрона атома водорода равно 1. Следовательно, орбитальное число равно 0, что соответствует шарообразной форме электронного облака, то есть единственный электрон атома водорода является s-электроном (размеры и форма электронного облака определяются распределением квадрата модуля пси-функции). S-орбиталь не расщепляется на подуровни, так как магнитное квантовое число при этом имеет одно значение ml
= 0. Спиновое квантовое число может принимать любое значение – либо
+1/2, либо -1/2. Таким образом, электронная формула атома водорода
1s1
.
В атоме второго элемента системы Менделеева, гелия, два электрона. Второй электрон, в соответствии с принципом наименьшей энергии, также располагается на 1s - уровне. Принцип Паули это разрешает, так как два электрона гелия имеют три одинаковых квантовых числа, но отличаются спиновым квантовым числом (у одного электрона - +1/2, у второго - -1/2). Электронная формула гелия 1s2
. Из неѐ видно, что электроны атома гелия находятся в спаренном состоянии.
В атоме водорода электрон находится в электромагнитном поле, которое создается только ядром. В многоэлектронных атомах на каждый электрон действует не только ядро, но и другие электроны, При этом электроны как бы сливаются в одно общее облако (это явление называется «гибридизацией
»). Решение уравнения Шрѐдингера в этом случае представляет сложную задачу, общим методом решения которой является одноэлектронное приближение. Для многоэлектронных атомов главное квантовое число n принимает значения от 1 до 7. У электронов второго от ядра энергетического уровня, например, имеет место следующая ситуация: n = 2; l
= 0 (s-электроны) и 1 (р-электроны); ml
=0 и ml
= -1, 0 и +1; ms
= +1/2 и -1/2. В соответствии с принципом Паули, максимальное количество электронов, которое может расположиться на втором энергетическом уровне, равно 8 (два s - электрона и 6 рэлектронов). Для s-электронов возможно только одно значение ml
, равное нулю, а для р – электронов - три значения, так как р-электронные облака ориентируются в пространстве по осям x, y и z, то есть происходит расщепление р- уровня на три подуровня.
Составим, для примера, электронную формулу элемента №8 (кислорода). Восемь электронов кислорода располагаются на двух ближайших к ядру орбитах следующим образом: 1s2
2s2
2p4
. Из четырѐх внешних электронов кислорода два являются неспаренными (выполняется правило Гунда). Электронная формула атома №5 (бора) выглядит следующим образом: 1s2
2s2
2p1
.
При n = 3 максимально возможное количество электронов на нем равно 18 (2 s-электрона + 6 р-электронов + 10 d-электронов). На четвертом уровне могут располагаться 32 электрона (2 s-электрона + 6 р-электронов + 10 d-электронов + 14 f-электронов). В общем случае, максимальное число электронов на энергетическом уровне равно 2n2
. Так, например, электронная формула элемента №15 (фосфора), принадлежащего III периоду, имеет вид: 1s2
2s2
2p6
3s2
3p3
3d0
.
Составляя электронные формулы атомов периодической системы, можно убедиться, что расположение их в периодической системе полностью соответствует электронному строению их атомов.
Если атом теряет один или несколько электронов, то превращается в положительно заряженный катион.
Аналогично, приобретая один или несколько «лишних» электронов, атом становится отрицательно заряженным анионом.
1.5. Проблема строения вещества
Проблема строения вещества
– это вопрос о том, существует ли всѐтаки конечный предел деления материи. В рамках атомномолекулярного учения пределом делимости материи считался атом. После того, как было установлено, что атом делим и были открыты электрон, протон и нейтрон, «первичными частицами материи» можно было посчитать именно эти частицы, которые были названы элементарными. И действительно, из протонов, нейтронов и электронов можно «построить» все известные химии элементы и даже создавать новые. Однако, начиная с середины ХХ века, было открыто более 400 различных элементарных частиц, в том числе нейтрино, мезоны, пионы, каоны, гипероны, резонансы, а также античастицы, например, «антиэлектрон», или позитрон. «Зоологический период», то количественный сбор информации о частицах в отсутствие приемлемой теории затянулся на десятки лет. Однако созданная в 60-е годы ХХ века теория элементарных частиц Гелл-Манна и Цвейга (которая теперь называется квантовой хромодинамикой
) строится на допущении наличия структуры у адронов, то есть у элементарных частиц, участвующих в так называемом сильном ядерном взаимодействии, самыми устойчивыми из которых являются протоны и нейтроны. Такие «суперэлементарные» частицы были названы кварками
. В переводе с немецкого языка слово «кварк» в переносном смысле значит «бессмыслица». И действительно, свойства кварков опрокидывают привычные физические представления: им приходится приписать дробный электрический заряд, равный 1/3 или 2/3 заряда электрона, и массу, почти в 10 раз превышающею массу протона и нейтрона, которые, как считается, состоят каждый из трех кварков. В 1969 году в экспериментах на линейном ускорителе в Стэндфордском университете были получены первые экспериментальные доказательства, подтверждающие существование кварков, а в 1994 г. американские исследователи сообщили об открытии «последнего», самого тяжелого из 36 кварков. До сих пор неизвестно, имеет ли структуру электрон; однако при ядерном бета-распаде он появляется «из нейтрона» в реакции, открытой итальянским физиком Э. Ферми: n
0  p
~ p
~
p
 n n
Нейтрон распадается на протон, электрон и антинейтрино, а протон – на нейтрон, позитрон и нейтрино. Следовательно, может существовать такое состояние вещества, когда электроны и кварки (из которых состоят нейтроны и протоны) представляют собой некую единую субстанцию. В таких обстоятельствах приходится возвращаться к проблеме строения вещества и вновь делать выбор между корпускулярной и континуальной концепциями. В современных представлениях о пределе делимости материи можно выделить три «условные концепции»:
«Философия матрешки наоборот»: предел делимости материи существует, но это должна быть не «самая маленькая», а «самая большая» частица, Ведь состоит же протон из трех кварков, каждый из которых в 10 раз больше его самого, следовательно, кварки, если у них есть структура, должны состоять из ещѐ более массивных частиц, те- в свою очередь из ещѐ более массивных, и т.д. Конечный предел деления материи в рамках этой концепции академик М.Марков ещѐ в 1965 г. назвал «максимоном» и определил ее массу как 10-5
г.
Концепция «суперструн», или «бутстрэпа» (от английского слова bootstrap
- шнурок от ботинка), или «ядерной демократии»: все состоит из всего (как протон из электрона и нейтрона, а нейтрон из протона и позитрона), и все частицы связаны «единым шнурком», как отверстия в ботинке. Такой «шнурок» некоторые считают особым уровнем существования вещества – «суперструной».
Обе эти концепции можно (хотя и весьма условно) назвать развитием корпускулярной концепции
строения материи.
Существует третья форма материи, или физический вакуум
, который способен рождать так называемые «виртуальные частицы» (копии реальных, время жизни которых менее 10-24
сек). Будучи созидательным, такой физический вакуум может претендовать на роль «первоматерии», «кочки и ухабы» которой образуют две другие формы материи – вещество и поле. Если это так, то проблему строения вещества следует решать в рамках континуальной концепции
. Из-за невероятной сложности современных представлений о строении материи некоторые ученые (так называемые «физики-диссиденты») предлагают вернуться к концепции эфира
, определяя ее, однако, несколько бессвязно: «эфир - это бесчастичная материя ньютоновского характера, носитель тепла, характеризует давление в газах, характеризует деффект масс, является основой элементарных частиц и любых веществ».
Дальнейшее развитие науки покажет, у какой концепции больше прав на существование.
1.6. Вещество на макроуровне
Поскольку вещество на микроуровне не имеет аналогов с наблюдаемыми нами явлениями макромира, можно назвать его микровеществом
. В обычной жизни человек имеет дело не с атомами и молекулами, а с веществом, находящимся в одном из четырех агрегатных состояний. В отличие от микровещества
(элементарных частиц, атомов и молекул) можно назвать вещество, с которым встречаемся в нашем макромире, макровеществом
. В зависимости от условий окружающей среды (в первую очередь от температуры и давления) одно и то же макровещество, например, вода, может находиться в различных агрегатных состояниях
. Различают газообразное, жидкое, твердое и плазменное
состояние вещества.
Можно применить системный подход и к макровеществу.
Так, система, состоящая из атомов и молекул вещества, которые находятся на больших (по сравнению с их собственными размерами) расстояниях друг от друга, является газом
и находится в газообразном состоянии,
Вследствие большой удаленности частиц газа друг от друга силы взаимодействия между ними пренебрежимо малы. Идеальный газ
– это система, элементы которой не взаимодействуют друг с другом. В реальном газе
между молекулами существуют заметные силы межмолекулярного взаимодействия. Паром
называют реальный газ, который находится в состоянии, близком к переходу его в жидкость. Молекулы в газах движутся хаотически (газ
в переводе с греческого языка означает хаос
). Главным видом движения молекул в газах является поступательное движение. При этом молекулы испытывают громадное число соударений (1020
соударений в секунду при комнатной температуре). Физико-химические свойства газовых систем определяются по правилу аддитивности (суммированием характеристик образующих их молекул). Свойства газовых систем определяются уравнением состояния Менделеева-Клайперона:
 mRT
(1.7) mRT
(1.7)
PV
M
 где P – давление; V – объем; m – масса газа; M - молекулярная масса газа; R - универсальная газовая постоянная (равная 8,3144103
Дж/кмольК, если измерять давление в Па, а объем в м3
); T - температура (в 0
Кельвина). где P – давление; V – объем; m – масса газа; M - молекулярная масса газа; R - универсальная газовая постоянная (равная 8,3144103
Дж/кмольК, если измерять давление в Па, а объем в м3
); T - температура (в 0
Кельвина).
При нагревании разреженных газов до высоких температур (десятков тысяч градусов) происходит ионизация молекул (от нейтральных атомов отщепляются электроны) и газ переходит в плазменное
состояние. Ионами
называют атомы или молекулы, которые приобретают или теряют один или несколько электронов и становятся заряженными частицами. Ионы в плазме способны к химическим реакциям, поэтому в плазме можно обнаружить такие экзотические с точки зрения химии частицы, как CH5
-
, H3
+
, He2
-
, Ne2
+
и т.п. Происходящие в плазме химические реакции
Когда вещество находится в конденсированном состоянии (жидком или твердом), расстояния между частицами малы, а силы взаимодействия между ними велики. Эти силы удерживают частицы вещества друг около друга, поэтому твердые и жидкие вещества, в отличие от газов, имеют постоянный при данной температуре объем. Силы, удерживающие частицы вещества, имеют электромагнитную
природу.
Если вещество состоит из атомов и не является металлом, его атомы связаны ковалентной связью. Если вещество - металл, то имеет место металлическая связь: часть электронов атомов металлов становится общей для всех атомов. В веществе, состоящем из ионов, последние удерживаются друг около друга электростатической ионной связью. В веществах с молекулярной структурой имеет место межмолекулярное
взаимодействие.
Силы межмолекулярного взаимодействия называют еще вандерваальсовыми
(по имени голландского учѐного Ван дер Вальса) силами. Эти силы слабее ковалентных сил, но проявляются на больших расстояниях. В их основе лежит явление поляризации
. Если вещество состоит из полярных молекул, например, молекул воды Н2
О или хлористого водорода НСl, то в конденсированном состоянии соседние полярные молекулы (диполи) ориентируются друг по отношению к другу противоположно заряженными полюсами, вследствие чего происходит их взаимное притяжение. Такой тип межмолекулярного взаимодействия называется ориентационным
. Ориентационное взаимодействие ослабевает с ростом температуры, так как ему препятствует тепловое движение молекул.
Если вещество состоит из неполярных, но способных к поляризации молекул, например СО2
, наблюдается возникновение наведенных, или индуцированных
диполей. Каждый атом создает вокруг себя электрическое поле, оказывающее воздействие на каждый атом соседней молекулы. Молекула поляризуется и в свою очередь поляризует соседние молекулы, в результате чего происходит притяжение молекул друг к другу. Индукционное взаимодействие имеет место и у полярных молекул, но оно значительно слабее ориентационного.
Движение электронов в атомах, а также колебание ядер и связанное с этим непрерывное изменение положения электронов вызывают появление мгновенных
диполей. В твердых телах и жидкостях мгновенные диполи возникают согласованно, причем ближайшие друг к другу участки соседних молекул оказываются заряженными противоположно, что приводит к их притяжению. Такое дисперсионное
взаимодействие имеет место во всех веществах, находящихся в конденсированном состоянии.
Относительная величина всех видов межмолекулярного взаимодействия зависит от полярности и поляризуемости молекул вещества. Чем больше полярность, тем больше ориентационные силы. Чем крупнее атомы (чем слабее связаны с ядром внешние электроны), тем значительнее дисперсионные силы. Индукционные силы почти всегда малы.
В твердом состоянии большинство веществ имеет кристаллическое
строение. Кристаллы отличаются от аморфных
соединений наличием упорядоченной кристаллической решетки
(См. Лекцию 15). Поэтому кристаллы, в отличие от аморфных твердых тел, имеют постоянную температуру плавления. Можно сказать, что структура кристаллов характеризуется дальним порядком
, а структура аморфных тел - ближним
.
Жидкое состояние
является промежуточным между твердым и газообразным состояниями. Способность жидкостей легко изменять свою форму, принимая форму сосуда, в который жидкость налита, говорит о том, что силы межмолекулярного взаимодействия в жидкостях не жестки. Однако жидкости практически несжимаемы (в отличие от газов), следовательно, межмолекулярные силы, хотя и не жесткие, тем не менее, значительны. По структуре жидкости подобны аморфным телам: в большинстве из них наблюдается ближний порядок
, то есть число ближайших соседей у каждой молекулы и их взаимное расположение приблизительно одинаковы во всем объеме данной жидкости. Первые теории жидкого состояния основывались на использовании аналогии с газовым состоянием. Современные теории жидкого состояния основаны на представлении о жидкости как о разупорядоченном твердом теле, в котором существует ближний порядок, в то время как дальний порядок, характерный для твердого тела, нарушен тепловым движением молекул. Жидкость можно рассматривать как тело, состоящее из очень большого числа беспорядочно ориентированных кристаллов микроскопических размеров.
1.7. Молекулярные системы
Все химические вещества делятся на простые (элементарные) и сложные
. Молекулы простых веществ состоят из одинаковых атомов (например, Сг, В, Н2
, О2
, Br2
). Простые вещества делятся на металлы и неметаллы
(металлоиды).
Сложные химические вещества делят на органические и неорганические
. Органическими веществами принято называть соединения углерода с водородом, азотом и некоторыми другими элементами; все остальные вещества называются неорганическими, или минеральными
.
Неорганические вещества делятся на классы либо по составу (двухэлементные, или бинарные, и многоэлементные; кислородсодержащие, азотсодержащие и т.п.) или по их функциональным признакам (кислоты, гидроксиды, восстановители, окислители и пр.).
К важнейшим бинарным соединениям относятся оксиды
(соединения элементов с кислородом); галогениды или галиды
(соединения элементов с галогенами, элементами подгруппы фтора); нитриды
(с азотом); карбиды (с углеродом); а также гидриды
(с водородом). Например, AgO - оксид серебра; Na F - фторид натрия; Mg3
N2
- нитрид магния; CaC2
- карбид кальция; СaH2
- гидрид кальция. Если менее электроотрицательный элемент может иметь переменную валентность, то после названия соединения валентность указывают в скобках римскими цифрами, например, Cu2
O - оксид меди (I), CuO - оксид меди (II), N2
O5
- оксид азота (V). Многие оксиды металлов имеют основной характер и реагируют с кислотами c образованием солей:
CuO +2HCl = CuCl2
+ H2
O.
Оксиды неметаллов имеют кислотный характер и реагируют с кислотами с образованием солей:
SO2
+ 2NaOH = Na2
SO3
+ H2
O.
Среди многоэлементных соединений одними из самых важных являются гидроксиды
- вещества, состоящие из гидроксильной группы ОН и металла. Основные
гидроксиды (например, КОН) проявляют свойства оснований (отщепляя в водном растворе гидроксильные группы), кислотные гидроксиды (например, HONO2
) в водном растворе ведут себя как кислоты, отщепляя ион водорода. Амфотерные основания (например, Zn(OH)2
) проявляют как те, так и другие свойства. Гидроксиды, образованные металлами первой группы (главной подгруппы) LiOH, NaOH, KOH, RbOH хорошо растворимы в воде, очень активны и носят тривиальное название щелочей
. Растворимы и некоторые гидроксиды металлов второй группы - кальция и бария. В зависимости от количества гидроксильных групп, входящих в состав гидроксидов, их делят на однокислотные
(NaOH), двукислотные
(Zn(OH)2
) и т.д.
Кислотами
называются соединения, способные в растворах отщеплять ионы водорода. Кислоты делятся на бескислородные (HCl, HF, H2
S, HCN и т.п.) и кислородсодержащие (H2
SO4
, H3
PO4
, HNO3
и пр.) В зависимости от числа входящих в молекулу кислоты атомов водорода кислоты подразделяют на одноосновные
, двухосновные, трехосновные
и т.д.
Соли
можно рассматривать как продукты замещения атомов водорода в кислоте атомами металлов (или группами других атомов, например, ионом аммония NH4
+
) или как продукты замещения гидроксогрупп в основном гидроксиде кислотными остатками.
При полном замещении получаются средние
(нормальные) соли (например, Na2
CO3
- карбонат натрия). При неполном замещении водорода, например, при реакции гидроксида с избытком кислоты, получаются кислые
соли (например, NaHCO3
- гидрокарбонат натрия), при неполном замещении гидроксила (при избытке гидроксида при получении соли) - основные соли (Mg(OH)Cl - хлорид гидроксомагния). Соли, образованные двумя металлами и одной кислотой, называются двойными
солями, или квасцами (например, KCr(SO4
)2
– сульфат калия-хрома, или хромокалиевые квасцы).
1.8. Химические формулы и расчѐты
Химические расчеты
основаны на том, что состав химических веществ можно выразить химическими формулами, а взаимодействия между веществами - химическими уравнениями.
Химическая формула
дает много сведений о веществе. Она показывает, из каких элементов состоит данное вещество и сколько атомов каждого элемента в его молекуле. Формула позволяет также рассчитать ряд величин, характеризующих вещество. Так, например, из формулы серной кислоты H2
SO4
видно, что молекула кислоты состоит из двух атомов водорода, одного атома серы и четырех атомов кислорода.
Для того чтобы определить формулу вещества, необходимо разложить определенное его количество на составные части (элементы), определить процентный состав каждого элемента и, зная атомную массу каждого элемента, проделать определенные расчеты. Так, серная кислота содержит 2 % водорода, 32 % серы и 64 % кислорода. Соответствующие атомные массы элементов равны 1, 32 и 16. Относительное содержание атомов каждого элемента (атомный фактор
) равно отношению процентного содержания элемента к его атомной массе. Отсюда следует, что в молекуле серной кислоты 2 атома водорода (2%:1), 1 атом серы (32%:32) и 4 атома кислорода (64%:16).
Относительную молекулярную массу[3]
рассчитывают по формуле вещества как сумму относительных атомных масс атомов, входящих в состав молекулы с учетом количества каждого атома. Так, молекулярная масса серной кислоты равна 2∙1+ 1∙32 + 4∙16 = 98.
Молем
называют количество вещества, в котором содержится число Авогадро молекул (6,02∙1023
). Это число обозначается символом NA
. Точное значение NA
определил не Авогадро, однако открытый им закон, который гласит, что масса 22,4 литра любого газа при нормальных условиях равна молекулярной массе газа, выраженной в граммах
. Отсюда вытекает вывод, что в таком количестве вещества содержится одинаковое число молекул. Этот вывод применим ко всем веществам, а не только к газам. Поскольку NA
молекул содержится в количестве вещества, равном массе молекулы (атома или иона), выраженном в граммах, то массу моля
(или молярную массу) любого вещества можно вычислить, определив молекулярную (атомную, ионную) массу и выразив еѐ в граммах
. Так, один моль водорода весит 2 г (водород в нормальных условиях существует в виде молекулы Н2
), один моль кислорода – 32 г (по той же причине), а один моль воды Н2
О– 18 г (1х2+16). Молярную массу
(массу моля) выражают в г/моль. Поскольку единицей массы атомов ныне считается 1/12 часть массы атома изотопа углерода С12
, то в 1971 году решением Генеральной конференции по мерам и весам в Париже было введено следующее понятие моля
.
Моль – единица количества вещества, равная количеству вещества системы, в которой содержится столько же структурных элементов (молекул, атомов, ионов, электронов, других частиц или специфицированных групп частиц), сколько атомов содержится в 0,012 кг нуклида 12
С (изотопа углерода с атомной массой 12)
.
Слово «моль» после числа и в заголовках таблиц не склоняют.
Эквивалентную массу
вещества вычисляют исходя из его молекулярной массы. Эквивалентная масса элемента равна его атомной массе, деленной на валентность элемента:
Э
 (1.8) (1.8)
Где Э- эквивалент элемента; А – его атомная масса, а В – валентность.
По закону эквивалентов
, вещества взаимодействуют друг с другом в количествах, пропорциональных их эквивалентам.
 m
1
Э
1
(1.9) m
1
Э
1
(1.9)
m
2 Э
2
где Э - эквивалент, m – масса соответствующего элемента.
Понятие «эквивалент»[4]
появилось гораздо раньше, чем атомномолекулярное учение. Например, на практике при смешении водорода с кислородом в соотношении 1:8 по массе вещества всегда реагируют без остатка (получается «хорошая» гремучая смесь). Поэтому при расчетах эквивалент кислорода принимается равным 8. Эквивалент водорода равен 1. Так как многие другие элементы вступают в реакции с кислородом и водородом, по закону эквивалентов (1.9) можно вычислить их эквиваленты. Эквивалентные массы элементов и сложных веществ могут принимать несколько значений, если элемент имеет переменную валентность, а вещество способно вступать в реакции разного типа. Величину, обратную валентности элемента, часто называют фактором эквивалентности F[5]
. У азота, например, для которого характерны валентности 1, 2, 3, 4 и 5, факторы эквивалентности равны 1, ½, 1/3, ¼ и
1/5.
Эквиваленты сложных соединений рассчитывают следующим образом:
Эквивалент кислоты Эк-ты
равен ее молекулярной массе М, деленной на основность кислоты (количество атомов водорода в ее молекуле) n(H):
 Э
к ты
М
к ты
(1.10) Э
к ты
М
к ты
(1.10)
n
(H
)
Фактором эквивалентности кислоты является величина, обратная
основности кислоты, или F
 . .
n
(H
)
Эквивалент гидроксида Эг-да
равен его молекулярной массе M, деленной на кислотность гидроксида (число гидроксильных групп в его молекуле) n(OH):
 Э
г да
М
г да
(1.11) Э
г да
М
г да
(1.11)
n
(ОH
)
Фактор эквивалентности гидроксида – это величина, обратная его
кислотности, или F
 . .
n
(OH
)
Эквивалент соли Эсоли
равен ее молекулярной массе M, деленной на валентность металла (В) умноженную на количество его атомов в молекуле (m):
М
соли
 Э
(1.12) соли
В т Э
(1.12) соли
В т
Фактор эквивалентности соли определяется соотношением: F
 . .
Молярной массой эквивалента (или молем эквивалента)
называют количество вещества, равное его эквивалентной массе (эквиваленту), выраженной в граммах. Так как моль-эквивалент составляет дробную часть моля или равен ему, его, в соответствии с рекомендациями ИЮПАК, рекомендуется называть просто молем с указанием фактора эквивалентности
. В общем случае, фактор эквивалентности вещества F равен молекулярной массе этого вещества M, делѐнной на массу его эквивалента Э:
 M
(1.13) M
(1.13)
F
Э
Любую реакцию можно выразить химическим уравнением
. Написав уравнение реакции, где в левой части приводятся формулы исходных веществ, а в правой – формулы продуктов реакции, следует соблюсти закон сохранения вещества, так как атомы не могут исчезать или появляться ниоткуда. Поэтому при составлении химического уравнения всегда уравнивают количества атомов в правой и левой его частях. При этом изменять формулы веществ нельзя.
Каждая формула в уравнении обозначает 1 молекулу вещества. Однако при количественном проведении реакции понятие «молекула» можно заменить понятием «моль», так как 1 моль любого вещества содержит одно и то число молекул - число Авогадро. Поэтому, зная молярные массы веществ - участников реакции и коэффициенты в уравнении, можно найти количественные соотношения между исходными веществами и продуктами реакции. Например, в уравнении 2NaOH + H2
SO4
= Na2
SO4
+ 2H2
O
2 молекулы гидроксида натрия вступают в реакцию с 1 молекулой серной кислоты. При этом получается 1 молекула сульфата натрия и 2 молекулы воды. Молекула – структурная единица микромира. Отсчитать молекулы «поштучно» с тем, чтобы в результате реакции все исходные вещества прореагировали целиком, без избытка одних и недостатка других, в макромире было бы невозможно. Но если в качестве «макромолекулы» взять число Авогадро (NA
), то для приведенной выше реакции справедлив следующий вывод: 2 NA
молекул гидроксида натрия вступают в реакцию с NA
молекул серной кислоты, при этом получается NA
молекул сульфата натрия и 2 NA
молекул воды, или 2 моль гидроксида натрия вступают в реакцию с 1 моль серной кислоты, при этом получается 1 моль сульфата натрия и 2 моль воды. Учитывая, что массы моля, или молярные массы, участвующих в реакции веществ равны соответственно 40, 98, 142 и 18 г/моль, уравнение можно прочесть так: 80г гидроксида натрия нацело взаимодействуют с 98 г серной кислоты с образованием 142 г сульфата натрия и 36 г воды. Моль является очень важной количественной характеристикой вещества, так как связывает структурные элементы микромира и макромира. Отвесив точно 1 моль любого вещества, мы всегда получаем строго определѐнное и точно известное (хотя и невообразимо большое) число молекул, а именно 6,02∙1023
. Исходя из уравнений реакции, по молярным соотношениям можно найти любую неизвестную величину, если известна масса хотя бы одного из участников реакции.
1.8. Процессы, происходящие в химических системах
Химические системы – это такие системы
, в которых идет химическая реакция. Химической системой является, например, содержимое любой колбы, в которой происходит превращение веществ, автоклава, где осуществляется синтез сложного органического вещества или ректификационной колонны на нефтеперерабатывающем заводе.
Процессы, происходящие в химических системах, можно разделить на две группы:
1. Превращения вещества, в результате которых изменяются структура или агрегатное состояние вещества, но химический состав его остается неизменным, называются фазовыми переходами
.
К ним относятся процессы плавления, испарения, кристаллизации, сублимации (переход твѐрдого вещества в газообразное состояние, минуя жидкую стадию) или растворения вещества. Например, фазовые превращения воды можно записать в виде следующих уравнений:
Н2
О(к) → Н2
О(ж) (плавление); Н2
О(ж) → Н2
О(г) (испарение);
Н2
О(ж) → Н2
О(к) (кристаллизация) Н2
О(к) → Н2
Ог (сублимация).
Растворение сульфата меди можно, например, представить следующим уравнением:
CuSO4
+ 5H2
O = CuSO4
∙5H2
O
2. Превращения, при которых изменяется химический состав и структура соединений, называются химическими реакциями.
Химические реакции можно разделить на следующие основные типы:
1) реакции соединения
(из простых веществ получаются более сложные)
2Cu + O2
= 2CuO;
H2
+ Cl2
= 2HCl
2) реакции разложения
(из сложного вещества получаются более простые)
2KMnO4
= K2
MnO4
+ MnO2
+ O2
; HCOOH = CO + H2
O;
3) реакции обмена
(молекулы взаимодействующих веществ обмениваются своими частями)
а) BaCl2
+ Na2
SO4
= BaSO4
+ 2NaCl; б) Na2
CO3
+ HCl = NaCl + CO2
+ H2
O; в) НCl + NaOH = NaCl + H2
O
;
Реакции обмена идут до конца, если в результате реакции образуется нерастворимое вещество (а), газ (б) или малодиссоциирующее соединение (в).
В противном случае реакция будет обратимой
, то есть будет идти как в прямом, так и в обратном направлении, а в числе продуктов реакции будут находиться и исходные вещества, образовывающие при обратном обмене (в ходе обратной реакции), например:
г) KCl + Na2
SO4
↔ K2
SO4
+ NaCl
Химическая система, в которой прямая и обратная реакция протекают с одинаковой скоростью, является равновесной
. По существу, все химические процессы являются обратимыми, хотя в некоторых (в частности, в приведенных выше процессах а, б
и в
) равновесие может быть сильно сдвинуто в сторону образования продуктов реакции. Изменяя условия, можно добиться сдвига равновесия в ту или иную сторону. Нерастворимые (или малорастворимые), газообразные и малодиссоциирующие вещества всегда принято записывать в виде молекулярной, а не ионной формулы. Растворимые вещества в растворе распадаются на положительно и отрицательно заряженные ионы (катионы и анионы соответственно) и их реакции можно записывать и в молекулярном, и в ионном виде. Например:
AgNO3
+ KCl = AgCl + KNO3 ,
или в полной ионной форме:
Ag+
+NO3
- + K+
+ Cl-
= AgCl + K+
+ NO3
-
,
или в краткой ионной форме:
Ag+
+ Cl-
= AgCl
Другой пример:
CH3
COONa + HCl = CH3
COOH + NaCl,
или в полной ионной форме:
CH3
COO-
+ Na+
+ H+
+ Cl-
= CH3
COOH + Na+
+ Cl-
или в краткой ионной форме:
CH3
COO-
+ H+
= CH3
COOH
4) окислительно-восстановительные
реакции (это такие реакции, в результате которых меняется степень окисления[6]
, или окисленность, входящих в молекулы элементов).
а) Zn + 2HCl = ZnCl2
+ H2
; б) 5K2
SO3
+ 2KMnO4
+ 3H2
SO4
= 6K2
SO4
+ 2MnSO4
+ 3H2
O
Кроме перечисленных выше основных типов реакций существуют и другие виды химических процессов, протекающих с изменением химического состава системы.
1.9. Вопросы и задания:
1. Дайте характеристику системно-структурному подходу к проблеме строения вещества.
2. Охарактеризуйте две концепции строения веществ
3. Опишите основные этапы становления атомно-молекулярного учения
4. Почему современную теорию строения вещества называют квантовой химией?
5. Сформулируйте принцип неопределенности Гейзенберга и принцип дополнительности Бора.
6. Сформулируйте «три закона квантовой механики» – правило Клечковского, принцип Паули и правило Гунда.
7. Что такое квантовые числа? Дайте им характеристику.
8. Для атома Х квадрат модуля пси-функции  = 4. Каковы значения главного квантового числа n для электронов этого атома? = 4. Каковы значения главного квантового числа n для электронов этого атома?
9. Напишите все квантовые числа для всех электронов атомов элементов с атомными номерами 3, 4, 5, 6, 7, 8, 9 и 10 (лития, бериллия, бора, углерода, азота, кислорода, фтора и неона).
10. Вычислите энергию, которую имеет электрон атома № 5, находясь на втором энергетическом уровне. Рассчитайте длину волны электрона, скорость которого равна 2∙103
м/сек.
11. Сколько элементов было бы в Периодической системе, если бы все квантовые ячейки на 7 ближайших к ядру орбитах были заполнены?
12. Какая электронная конфигурация соответствует хлорид-иону Cl-
; иону О2-
(оксид-иону)?
13. Сколько неспаренных электронов содержит атом углерода в основном состоянии?
14. Определите молярную массу 0,327∙10-3
л газа при 13о
С и давлении
1,040∙105
Па, если масса газа в данных условиях равна 0,828∙10-3
кг.
15. Напишите формулы оксида индия, гидрида титана, нитрида бора, карбида кальция, гидроксида рубидия и селеновой кислоты.
16. Что образуется при взаимодействии оксида серы (IV) с избытком раствора NaOH: гидросульфит натрия, гидросульфит натрия и вода, гидросульфат натрия; сульфат натрия и вода?
17. Сколько хлорного железа нацело прореагирует с 12 г гидроксида натрия в реакции FeCl3
+ 3 NaOH = Fe(OH)3
+ 3NaCl? Сколько при этом образуется
гидроксида железа (III) хлорида натрия?
18. Какие из приведенных ниже реакций осуществимы в водном растворе? NaNO3
+ HCl = NaCl + HNO3
; Ba(NO3
)2
+ 2NaOH = 2NaNO3
+ Ba(OH)2
;
Fe2
(SO4
)3
+ 6HNO3
= 3Fe(NO3
)2
+ 3H2
SO4
; CuSO4
+ 2KOH = K2
SO4
+ Cu(OH)2
; ВаSO4
+ 2HCl = BaCl2
+ H2
SO4
; CaCl2
+ 2NaNO3
= Ca(NO3
)2
+ 2
NaCl; FeCl3
+ 3 NaOH = Fe(OH)3
+ 3NaCl; K2
SO4
+ 2HCl = H2
SO4
+ 2KCl
19. Определите молярную массу следующих веществ: NH4
ОН, H2
SO4
, KOH, Fe2
(SO4
)3
, Н2
О, К2
Cr2
O7
, CH3
COOH.
20. Определите молярную массу эквивалента для следующих веществ:
H2
SO4
, Cu(OH)2
, Al2
(SO4
)3
; HNO3
; Na2
SiO3
; NaOH
Лекция 2. Растворы
2.1. Раствор как система
Растворами называются гомогенные системы переменного состава
. Компонент, содержание которого в растворе больше содержания других компонентов, называется растворителем
. Остальные компоненты раствора называются растворенными веществами
. Растворитель, в отличие от растворенного вещества, при образовании раствора сохраняет свое агрегатное состояние.
Существуют газовые, твердые и жидкие растворы
.
В газовом
растворе частицы слабо взаимодействуют друг с другом, поэтому газовые растворы можно считать физической смесью компонентов. Примером газового раствора является воздух, где растворителем можно считать азот (80%), а растворенными веществами – кислород (около 20%), диоксид углерода, пары воды и инертные газы (до 1%). При невысоких давлениях газы смешиваются друг с другом в любых соотношениях, а при повышении давления растет растворимость в газах жидких и твердых веществ. Например, в пар высокого давления из жидкого раствора переходят соли, оксиды кремния и другие вещества.
Твердые
растворы – это сложные системы, в которых атомы различных элементов расположены в общей кристаллической решетке
. Все кристаллические твердые тела способны образовывать твердые растворы; пределы растворимости растворяющегося компонента обычно узкие, хотя имеются системы с полной взаимной растворимостью, например, Cu-Ag, Ag- Au.
К жидким
растворам относятся растворы газов, жидкостей и твердых веществ в жидких растворителях. Различают водные и неводные
растворы.
Однородность растворов, отличающая их от механических смесей, в то же время делает их похожими на химические соединения. При растворении некоторых веществ выделяется или поглощается теплота, что свидетельствует о химическом взаимодействии между молекулами растворителя и растворяемого вещества. Однако растворы отличаются от химических соединений тем, что их состав может меняться в широких пределах и что в растворе можно обнаружить свойства его отдельных компонентов.
При растворении многих веществ их молекулы (или ионы) связываются с молекулами растворителя, образуя соединения, называемые сольватами
или, в случае водных растворов, гидратами
. Гидраты, как правило, неустойчивы и при выпаривании раствора разлагаются. Однако, некоторые гидраты настолько прочны, что при выделении растворенного вещества из раствора вода входит в состав молекулы вещества. Такие вещества называются кристаллогидратами
, а содержащаяся в них вода – кристаллизационной. Например, кристаллогидрат сульфата меди (медного купороса) имеет состав CuSO4
∙5H2O
, кристаллогидрат сульфата натрия (глауберова соль) Na2
SO4
∙10H2
O
, кристаллогидрат карбоната натрия (соды) Na2
CO3
∙10H2
O
, кристаллогидрат бората натрия (буры) Na2
B2
O7
∙10H2
O
, кристаллогидрат тиосульфата натрия Na2
S2
O3
∙5H2
O
.
2.2 Растворение и растворимость
Растворение кристалла в жидкости происходит за счет собственного колебательного движения молекул растворяемого вещества, а с другой стороны, вследствие силы притяжения молекул растворителя.
Перешедшие в раствор молекулы, ударяясь о поверхность кристалла, вновь притягиваются к нему и опять входят в состав кристалла. В некоторый момент скорость растворения становится равной скорости кристаллизации вещества. При этом устанавливается динамическое (подвижное) равновесие
: в единицу времени растворяется и кристаллизуется одинаковое количество молекул растворенного вещества. Такой раствор называется насыщенным
.
Растворимостью
называется способность вещества растворяться в том или ином растворителе. Мерой растворимости служит содержание вещества в его насыщенном растворе. Чаще всего растворимость выражают в граммах растворенного вещества на 100 г растворителя при данной температуре. Эта величина называется коэффициентом растворимости
. Если в 100 г воды растворяется более 10 г вещества, последнее называется хорошо растворимым
соединением; если менее 1 г – малорастворимым
; а если в раствор переходит менее 0,01 г вещества, его называют практически нерастворимым
. При растворении твердого тела растворение прекращается, когда образуется насыщенный раствор, то есть когда между молекулами растворяемого вещества (соли) в кристалле и ионами его в растворе установится равновесие:
СаSO4
↔ Ca2+
+ SO4
2-
;
Константой равновесия[7]
данного процесса называется соотношение:
K
 (2.1) (2.1)
[CaSO
4
]
Знаменатель дроби – концентрация твердой соли – представляет собой постоянную величину, которую можно ввести в константу равновесия и обозначить ПР. Тогда получаем:
[Ca2+
] [SO4
2-
] = ПР;
Таким образом, в насыщенном растворе электролита произведение концентраций его ионов есть величина постоянная. Эта величина характеризует способность электролита растворяться и называется его произведением растворимости (ПР)
. Численное значение ПР легко найти, зная растворимость электролита. Например, растворимость сульфата кальция СаSO4
при 200
С равна Р = 1,5∙10-2
моль/л. Это значит, что в растворе концентрация каждого иона (кальция и сульфат - иона) также равна 1,5∙10-2
моль/л. Следовательно, ПР этой соли равно (1,5∙10-2
)2
= 2,25∙10-4
. Если электролит содержит два или несколько одинаковых ионов, их концентрации при вычислении ПР должны быть возведены в соответствующую степень. Например, для йодистого свинца PbI2
ПР = [Pb2+
]∙[J-
]2
, а для фосфорнокислого серебра Ag3
PO4
ПР = [Ag]3
∙[PO4
]. В общем случае, когда
ПР = [Km+
]n
∙[An-
]n
, где К – катион, а А – анион, растворимость иона рассчитывается из соотношения:
P
 (2.2) (2.2)
Где Р – растворимость, ПР – произведение растворимости, а m и n – количество катионов и анионов в молекуле соли соответственно. Чем меньше растворима соль, тем меньшее значение имеет ее произведение растворимости. В тот момент, когда произведение концентраций ионов данного трудно растворимого соединения достигает величины его произведения растворимости при данной температуре, раствор становится насыщенным, и при дальнейшем увеличении концентрации ионов происходит выпадение осадка, то есть:
Если ПР < [Km+
]n
∙[An-
]n
, происходит выпадение осадка Если ПР = [Km+
]n
∙[An-
]n
, раствор и осадок находятся в равновесии Если ПР > [Km+
]n
∙[An-
]n
, происходит растворение осадка
Чем меньше значение ПР, тем быстрее данное трудно растворимое вещество выпадает в осадок. Из двух малорастворимых соединений в первую очередь будет осаждаться то, ПР которого меньше.
В большинстве случаев на практике используют ненасыщенные растворы; при этом растворы с относительно высоким содержанием растворенного вещества называют концентрированными
, а с низким – разбавленными
.
2.3 Способы выражения концентрации растворов
Состав раствора (содержание в нем растворенного вещества) может выражаться различными способами, как при помощи безразмерных единиц (долей или процентов), так и через размерные величины – концентрации. В химической практике применяют следующие способы выражения концентрации растворов:
Массовая доля, или процентная концентрация[8]
– отношение (обычно выраженное в процентах
) массы растворенного вещества к массе раствора.
 m
1
100 m
1
100
(2.3) m
2
Где ω – массовая доля, m1
– масса растворѐнного вещества, а m2
– масса раствора. Например, 15% по массе водный раствор хлорида натрия – это такой раствор, который содержит в 100 г (или других единицах массы) 15 г (или других единиц массы) хлорида натрия и 85 г (или других единиц массы) воды.
Молярная доля, или мольная доля
– отношение количества молей растворенного вещества или растворителя к сумме молей всех составляющих раствор веществ. Для бинарной системы растворитель - растворенное вещество мольная доля растворенного вещества (N2
) равна N2
= n2
/(n1
+ n2
),
а мольная доля растворителя (N1
) равна N1
= n1
/(n1
+ n2
),
где N - мольная доля, n1
– число молей растворителя, n2
– число молей растворѐнного вещества.
Например, 100 г 36% раствора глюкозы (С6
H12
O6
) содержит 36 г глюкозы и 64 г воды. Определяем количество молей глюкозы n2
и воды n1
:
n2
= 36/180 = 0,20 моль; n1
= 64/18 = 3,56 моль. Отсюда,
N2
= n2
/(n1
+ n2
) = 0,20/(0,20+3,56) = 0,053; N1
= n1
/(n1
+ n2
) = 3,56/(0,20+5,56) = 0,947.
Сумма молярных долей всех компонентов раствора всегда равна единице.
Молярная концентрация
вещества (М)
, - это отношение количества молей растворенного вещества к объему раствора, выраженному в
литрах, или
M
 (2.4) (2.4)
M
mol
V
где m
– масса растворенного вещества в г; Мmol
– его молярная масса в г/моль, а V
– объем раствора в литрах.
Другими словами, молярная концентрация - это количество молей растворенного вещества в одном литре раствора. Так, раствор серной кислоты с молярной концентрацией 2М содержит 2 моль серной кислоты (или 2∙98 г) в одном литре раствора, то есть молярная концентрация его равна 2 моль/л.
Моляльная концентрация, или моляльность (m) –
это отношение количество молей растворенного вещества к массе растворителя.
m
 M
(2.5) M
(2.5)
M
mol
m
2
где m1
– масса растворенного вещества; Мmol
– его молярная масса, а m2
– масса растворителя, кг.
Например, водный раствор серной кислоты с моляльной концентрацией 2m
содержит 2 моль (или 2∙98 г) серной кислоты на 1 кг растворителя (воды), то есть моляльность раствора равна 2моль/кг (H2
O). В отличие от молярности, моляльность не изменяется с изменением температуры.
Молярная концентрация эквивалента (эквивалентная концентрация, или нормальная концентрация, или нормальность), обозначаемая буквами N или н - это отношение количества эквивалентов растворенного вещества к объему раствора в литрах
m
 N
(2.6) N
(2.6)
ЭV
где m – масса растворѐнного вещества в г; Э – моль-эквивалент растворѐнного вещества моль, а V – объѐм раствора в л.
Другими словами, нормальность – это количество моль-эквивалентов растворенного вещества в одном литре раствора. Например, раствор серной кислоты с концентрацией 2н содержит 2 эквивалента серной кислоты в одном литра раствора, то есть нормальность раствора равна 2г-экв/л. Зная, что эквивалент серной кислоты равен ее молекулярной (молярной) массе, деленной на основность кислоты, можно рассчитать молярность однонормального раствора (0,5 моль/л) и массу кислоты в 1 л раствора (49 г/л).
В соответствии с рекомендациями Международного союза по теоретической и прикладной химии (ИЮПАК), молярной концентрацией
раствора ныне называется количество молей
растворѐнного вещества в 1 килограмме растворителя
, то есть такая концентрация, которая раньше называлась моляльной. Употребление таких терминов, как моляльная концентрация, моляльность, нормальная концентрация и нормальность ИЮПАК не рекомендует
. Тем не менее, на практике гораздо удобнее готовить объѐмные растворы, которые не требуется взвешивать. Так, растворы нормальной концентрации широко применяются в титримерическом анализе, где необходимо точно знать объемы растворов полностью прореагировавших веществ. Поскольку все вещества реагируют пропорционально их эквивалентам, то для растворов справедливо соотношение:
   NVconst
,
или N
1
V
1
N
2
V
2
,
а также N
2
N
1
V
1
(2.7) NVconst
,
или N
1
V
1
N
2
V
2
,
а также N
2
N
1
V
1
(2.7)
V
2
где N – нормальная концентрация в моль-экв/л; а V – объѐм раствора в литрах.
Зная объемы прореагировавших веществ и нормальную концентрацию одного из них, можно определить неизвестную концентрацию и содержание вещества в растворе. В объѐмном анализе также широко используется термин «титр».
Титром раствора (Т)
называется масса вещества в граммах, содержащаяся в 1 мл (см3
) раствора. Титр выражается в г/мл. Например, в 1 мл раствора соляной кислоты с титром Т = 0,02 г/мл содержится 0,02 г HCl. Титр связан с молярной концентрацией эквивалента соотношением
 Э N
(2.8) Э N
(2.8)
Т
1000
где Э – эквивалентная масса растворенного вещества, N – нормальная концентрация раствора (молярная концентрация эквивалента).
2.4. Электролитическая диссоциация
Многие растворы, особенно водные растворы солей, кислот и оснований отличаются от других растворов тем, что проводят электрический ток
(следовательно, содержат заряженные частицы), а также другими физическими свойствами (например, более высоким осмотическим давлением, повышением температуры кипения и понижением температуры замерзания в большей степени, чем это отвечает их концентрации). Причиной указанных явлений является электролитическая диссоциация, имеющая место в растворах солей, оснований и кислот, которые вследствие этого называются электролитами.
Теорию электролитической диссоциации
разработали С. Аррениус, И.А. Каблуков и Д.И. Менделеев.
При растворении кристаллов с ионной структурой и полярных молекул поляризация ионов и молекул молекулами полярного растворителя (воды) в сочетании с непрерывным колебательным движением атомов приводит к распаду полярной молекулы на ионы. Так, например, при растворении в воде хлористого водорода образуется хлористоводородная кислота, а также имеет место следующий процесс: HCl = H+
+ Cl-
;
Аррениус отметил, что на ионы диссоциирует только часть молекул электролита, и ввел понятие степени диссоциации
. Степенью диссоциации электролита называется отношение числа его молекул, распавшихся на ионы, к общему числу молекул
. Все электролиты можно разделить на сильные и слабые. Сильные электролиты в водных растворах диссоциируют практически нацело. Слабые электролиты диссоциируют только частично, в растворе устанавливается динамическое равновесие между недиссоциированными молекулами и ионами. К сильным электролитам относятся почти все растворимые соли; из важнейших кислот и оснований к ним относятся HNO3
, H2
SO4
, HClO4
, HCl, HBr, HJ, NaOH, KOH, Ba(OH)2
и Ca(OH)2
.
К слабым электролитам относится большинство органических кислот, в том числе СН3
ООН
, а из важнейших неорганических соединений к ним принадлежат кислоты угольная H2
CO3
,
сероводородная H2
S,
циановодородная HCN,
кремниевая H2
SiO3
и гидроксид аммония NH4
OH
. Степень диссоциации обозначают греческой буквой альфа (α) и выражают либо в долях единицы, либо в процентах. Так, для 0,1н (децинормального) раствора уксусной кислоты степень диссоциации равна 0,0013 (или 1,3%), а для 0,1рн раствора синильной кислоты 10-4
(или 0,01%). Для слабых электролитов степень диссоциации выражают при помощи константы диссоциации. Константой диссоциации слабого электролита - например, уксусной кислоты, диссоциирующей по уравнению:
СН3
СООН = СН3
СОО-
+ Н+
- называют соотношение:
 [H
] [CH
3
COO
] 2 10 5
(2.9) [H
] [CH
3
COO
] 2 10 5
(2.9)
K
д
[CH
3
COOH
]
Где Кд – константа диссоциации, а в квадратных скобках приведены молярные концентрации ионов водорода, ацетат-аниона и уксусной кислоты.
Многоосновные кислоты диссоциируют ступенчато. Например, диссоциация угольной кислоты происходит в две ступени: 1) H2
CO3
= H+
+ HCO3
-
; 2) HCO3
-
= H+
+ CO3
2
-
.
Диссоциация по первой ступени характеризуется константой
 [H
] [HCO
] 7
, [H
] [HCO
] 7
,
K
1
4,5 10 [H
2
CO
3
]
а диссоциация по второй ступени – константой
 K
2
[H
] [CO
32 ] 4,7 10 11. K
2
[H
] [CO
32 ] 4,7 10 11.
[HCO
3
]
Суммарная константа диссоциации
 K
[H
]2 [CO
32 ] 7 2,11 10 17 K
[H
]2 [CO
32 ] 7 2,11 10 17
[H
2
CO
3
]
Величины К1
, К2
и К связаны соотношением: К = К1
К2
, причем К1
всегда меньше К2
. Таким же образом связаны константы ступенчатой диссоциации оснований.
Если обозначить концентрацию электролита на два иона через С, а степень его диссоциации через α, то концентрация каждого из ионов будет Сα, а концентрация недиссоциированных молекул С(1- α). Тогда уравнение для константы диссоциации приобретает вид:
  C C
K
или K
Это уравнение выражает закон разбавления Оствальда
. Для очень слабых электролитов, где α много меньше 1, ее величиной в знаменателе можно пренебречь. При этом уравнение приобретает вид:
K
 C
,
или C
,
или  K
(2.10) K
(2.10)
Из этого уравнения наглядно видно, что степень диссоциации возрастает при разбавлении раствора
. Уравнение дает возможность вычислять степень диссоциации электролита при известной константе диссоциации.
2.5. Ионная сила раствора и коэффициент активности
В растворах сильных электролитов молекулы почти полностью диссоциируют на ионы, поэтому количество ионов велико и расстояния между ними малы. При этом существенную роль играют силы притяжения между ионами, поскольку каждый ион окружен так называемой ионной атмосферой. «Тормозящее» действие ионной атмосферы приводит к понижению степени диссоциации; в связи с этим определяемая (по электрической проводимости) степень диссоциации
называется кажущейся
. Для оценки состояния ионов в растворах сильных электролитов вместо понятия «концентрация» пользуются понятием «активность»[9]
. Активность иона равна его концентрации С, умноженной на коэффициент активности
f; то есть
a
 fc
(2.11) fc
(2.11)
В концентрированных растворах коэффициент активности обычно меньше единицы, а с разбавлением раствора приближается к этой величине. Природа ионов мало влияет на коэффициент активности. Можно считать, что коэффициент активности зависит только от заряда иона и ионной силы раствора
.
Ионной силой раствора называют полусумму произведений концентраций всех ионов в растворе на квадрат их заряда.
I = ½(C1
z2
1
+ C2
z2
2
+ … + Cn
z2
n
)
(2.12) где С – концентрация иона в моль/л, а z – заряд иона.
Например, в растворе, содержащем 0,1 моль/л хлорида натрия и 0,1 моль/л хлорида бария, концентрация ионов натрия с зарядом +1 равна 0,1 моль/л; концентрация ионов бария с зарядом +2 равна 0,1 моль/л, а общая концентрация хлорид - ионов с зарядом –1 равна (0,1 + 2∙0,1).
Таким образом, ионная сила этого раствора равна:
I = ½ [0,1∙12
+ 0,1∙22
+ 0,3∙ (-1)2
] = 0,4
.
Коэффициент активности и ионная сила раствора связаны соотношением:
 lg f
0.5Z
2
I
(2.13) lg f
0.5Z
2
I
(2.13)
Где f – коэффициент активности, Z – заряд иона, а I ионная сила раствора
Активность электролита равна произведению активности его ионов a = а+m
∙ а—n
(2.14)
где а+
- активность катиона; а-
- активность аниона; m – количество катионов; n – количество анионов в молекуле.
Значения термодинамических констант диссоциации
сильных кислот можно получить, заменяя концентрации ионов их активностями.
Это дает возможность сравнивать, например, сильные кислоты. Так, для азотной, бромистоводородной, иодистоводородной, марганцевой, серной и соляной кислот термодинамические константы диссоциации соответственно равны 46,3; 109
; 1011
; 200; 1000 (К1
) и 10-2
(К2
); 107
.
2.6. Физические свойства растворов
Используя физические свойства растворов, можно определить молекулярную массу растворенного вещества. Такими свойствами являются осмотическое давление раствора, понижение температуры замерзания и повышение температуры кипения раствора при растворении вещества.
Если поместить в сосуд концентрированный раствор какого-либо вещества, например, сахара, а сверху осторожно налить менее концентрированный раствор его, то вначале молекулы сахара и воды будут распределены в объеме раствора неравномерно. Однако через некоторое время молекулы воды и сахара равномерно распределятся во всем объеме раствора вследствие диффузии
молекул растворенного вещества концентрированного раствора в разбавленный, а молекул растворителя – из разбавленного раствора в концентрированный. Если между двумя растворами поместить полупроницаемую перегородку, через которую растворитель может проходить, а растворенное вещество не может, то выравнивание концентраций будет происходить только за счет перемещения молекул воды. Объем раствора будет при этом увеличиваться, а концентрация сахара в нем будет уменьшаться. Такая односторонняя диффузия через полупроницаемую перегородку называется осмосом
. Давление, создаваемое при увеличении объема раствора до прекращения диффузии молекул растворителя через перегородку, называется осмотическим давлением раствора
. Явление осмоса имеет место в природе. Оболочки клеток растений проницаемы для воды, но почти непроницаемы для веществ, растворенных во внутриклеточной жидкости; вода, проникая в клетки, создает в них избыточное давление, растягивая оболочки клеток и поддерживая их в напряженном состоянии. Осмос является причиной питания клеток и многих других явлений. В промышленности осмос используют для опреснения воды. Солевой раствор (морскую воду) отделяют полупроницаемой мембраной от пресной воды и подвергают более высокому давлению, чем осмотическое давление раствора (это так называемый обратный осмос
). В результате часть воды переходит в фазу пресной воды. Сконцентрированный солевой раствор заменяют свежими порциями подлежащей опреснению воды. В 1886 году голландский химик Я.Г. Вант Гофф показал, что для разбавленных растворов неэлектролитов
зависимость осмотического давления от концентрации и температуры выражается следующим законом (Вант Гоффа):
PCRT
(2.15)
где Р – осмотическое давление раствора; С – его молярная концентрация; Т – абсолютная температура; R – универсальная газовая постоянная, равная 8,314 Дж/моль∙К,
или PV
mRT
(2.16) M
где m – масса растворенного вещества; M – молекулярная масса растворенного вещества; V – объем раствора, а R – универсальная газовая постоянная, равная 8,314 кДж/моль∙К.
Пользуясь (2.16), можно по величине осмотического давления определить неизвестную молекулярную массу растворенного вещества. Например, если осмотическое давление раствора, в 250 мл которого содержится 3 г сахара при температуре 120
С, равно 83,14 кПа, то можно определить молекулярную массу сахара: 83,14∙0,25 = 3∙8,314(373+12)/М; отсюда, М = 342 г/моль.
Индивидуальные вещества характеризуются строго определенными значениями температуры при переходе из одного агрегатного состояния в другое (температурой фазового перехода). Так, вода при атмосферном давлении (101,3 кПа) кипит при 1000
С, а замерзает при 00
С. Присутствие растворенного вещества повышает температуру кипения раствора и понижает температуру его замерзания. В 1887 году французский химик Рауль установил закон, в соответствии с которым для разбавленных растворов неэлектролитов повышение температуры кипения и понижение температуры замерзания определяются следующим соотношением:
 (2.17) (2.17)
где E – эбуллиоскопическая константа и К – криоскопическая константа; mв
– масса растворѐнного вещества, г; М – молекулярная масса растворѐнного вещества; mр
– масса растворителя, г. Эбуллиоскопическая и криоскопическая константы зависят только от природы растворителя, но не зависят от природы растворенного вещества. Например, для воды К равна 1,86, а Е равна 0,52; для бензола К=5,07; Е=2,6.
2.7. Вопросы и задания:
1. Какие способы выражения концентрации растворов вы знаете?
2. Как изменяется степень диссоциации при разбавлении раствора?
3. Растворимость фосфорнокислого серебра Ag3
PO4
в воде при 200
С равна 0,0065 г/л. Рассчитайте значение произведения растворимости этого соединения.
4. Рассчитайте массовую долю соли в растворе, полученном при смешении 150 г 2% раствора и 350 г 4% раствора.
5. Какова молярная концентрация 1 л раствора соляной кислоты с массовой долей 36,6%, плотность которого равна 1,18 г/мл.
6. Определите массу осадка, образующегося при смешении 100 мл 0,1М раствора FeCl3
и 150 мл 0,2М раствора NaOH.
7. Чему равна массовая доля фосфата калия в растворе, полученном при растворении 0,5 моля соли в 124 мл воды?
8. Определите ионную силу раствора хлорида кальция СаCl2
и коэффициенты активности ионов кальция и хлора в водном растворе, содержащем 0,555 г соли в 500 мл раствора.
9. Вычислите осмотическое давление раствора, содержащего в 1,4 л 63 г глюкозы C6
H12
O6
при 00
С.
10. Определите температуру кипения и замерзания раствора, содержащего 1 г нитробензола C6
H5
NO2
в 10 г бензола. Эбуллиоскопическая и криоскопическая константы бензола соответственно равны 2,57 и 5,1 0
С. Температура кипения чистого бензола равна 80,2 0
С, температура его замерзания равна +5,4 о
С.
Лекция 3. Дисперсные системы
3.1. Классификация дисперсных систем
При растворении вещества в растворителе можно получить системы с частицами разного размера. Если вещества присутствуют в растворе в виде молекул атомов или ионов, то такие системы называются истинными растворами.
Если же мы разобьем нерастворимое в воде вещество на мельчайшие, но не молекулярные или ионные частицы, то можем получить дисперсную систему
с различными размерами частиц. Дисперсные системы не могут быть получены самопроизвольно. Для этого нужно совершить определенные действия, связанные с затратой энергии. Дисперсные частицы можно получить либо дроблением крупных частиц, либо объединением мелких частиц (молекул, атомов или ионов) в более крупные агрегаты. Первый метод называется диспергированием,
а второй конденсацией
.
Для получения дисперсных систем первым способом используются шаровые мельницы – полые вращающиеся цилиндры, содержащие некоторое количество стальных или керамических шаров. При вращении цилиндра эти шары перекатываются, дробя и истирая измельчаемый материал. В шаровых мельницах получают порошки, цемент, густотертые краски и пр. Для более тонкого измельчения вещества используют так называемые коллоидные мельницы, а также ультразвуковые методы.
К методам получения дисперсных систем методом конденсации относятся конденсация паров в газовой фазе; конденсация паров при пропускании их через холодную жидкость; замена растворителя; а также химические методы, когда дисперсные системы образуются при получении малорастворимых соединений в результате химических реакций, проведенных в определенных условиях, когда вместо осадка получается дисперсная система.
Дисперсные системы являются гетерогенными системами. Они состоят из сплошной непрерывной фазы – дисперсионной среды
и находящихся в этой среде раздробленных частиц различного размера и формы – дисперсной фазы
.
Дисперсные системы классифицируют по дисперсности (степенью дисперсности называется величина, обратная размеру частицы) на грубодисперсные системы
(с размерами частиц от 1 до 10-2
см) и коллоидные растворы
(с размерами частиц от 10-2
до 10-7
см). Частицы грубодисперсных систем различимы в обычный микроскоп, задерживаются бумажным фильтром и расслаиваются при стоянии. Частицы коллоидных растворов проходят через бумажный фильтр, невидимы в обычный микроскоп и не изменяются при стоянии.
Истинные растворы
, о которых шла речь в предыдущей лекции, отличаются от дисперсных систем (коллоидных растворов) размерами частиц растворенного вещества. В растворах неэлектролитов частицы растворенного вещества являются молекулами; в растворах электролитов – ионами. Размеры этих частиц порядка 10-8
см и менее. Часто трудно отличить коллоидные системы от истинных растворов только по таким внешним признакам, как прозрачность и однородность. Классификация систем по дисперсности не дает полного представления о свойствах систем, поэтому их дополнительно классифицируют по агрегатному состоянию дисперсной фазы и дисперсионной среды.
Существует восемь вариантов сочетания агрегатных состояний (см.
Таблицу 3.1):
К системам с жидкой дисперсионной средой относятся лиозоли
, или коллоидные растворы металлов (золота и серебра), взвеси, суспензии; эмульсии (молоко, майонез, маргарин, эмульсолы); пены.
К системам с твердой дисперсной средой относятся твердые золи (цветное стекло, сплавы, драгоценные камни); твердые эмульсии, или гели, представляющие собой вкрапления капелек жидкости в твердое тело (жемчуг, опал, алюмогель, силикагель); твердые пены (пемза, туф, пенобетон, пенопласт и др.) К системам с газообразной дисперсной средой относятся пыль, дым, туман.
Таблица 3.1. Виды дисперсных систем
| Дисперсионная среда
|
Дисперсная фаза
|
| Твердое тело
|
Жидкость
|
Газ
|
| Жидкость
|
Суспензия; коллоидная система (золь)
|
Эмульсия
|
Пена
|
| Твердое тело
|
Сплавы, минералы
|
Гель, твердая эмульсия
|
Твердая пена, пористые тела
|
| Газ
|
Дым, пыль
(аэрозоль)
|
Туман (аэрозоль)
|
-
|
3.2. Коллоидные растворы
Дисперсные системы, в которых дисперсионной средой является жидкость, а размер частицы дисперсной фазы лежат в пределах от 10-2
до 10-7
см, называются золями,
или коллоидными растворами.
Коллоидные системы широко распространены в природе, а также играют значительную роль во многих производственных процессах
(водоподготовка, очистка сточных вод).
Структурной единицей коллоидной системы является мицелла (см. Рис.3.1)
, а входящая в неѐ частица дисперсной фазы носит название коллоидной частицы
. Коллоидная частица состоит из ядра
, которое образуют частицы малорастворимого вещества
. Ядро адсорбирует из дисперсной среды ионы родственной ядру природы. Эти ионы называются зарядообразующими
, или потенциалопределяющими
, их химическая природа близка природе ядра коллоидной частицы. Ядро коллоидной частицы с адсорбированными зарядообразующими ионами притягивает к себе из дисперсионной среды ионы противоположного знака (противоионы). В состав коллоидной частицы входит только часть имеющихся в системе противоионов (их называют связанными), которые образуют адсорбционный слой.
Другая часть противоионов остается в дисперсионной среде, в жидкой фазе, образуя диффузный слой
. Эти противоионы называются свободными. Противоионы адсорбционного и диффузного слоев находятся в состоянии динамического равновесия
адсорбции – десорбции.
Рис. 3.1. Строение мицеллы йодида серебра

Ядро совместно с адсорбционным слоем противоионов является коллоидной частицей.
Электрический заряд коллоидной частицы равен сумме зарядов потенциалопределяющих ионов и ионов адсорбционного слоя. Этот заряд, возникающий на коллоидной частице, приводит к возникновению потенциала, который называется ξ (дзета)- потенциалом
. Коллоидная частица ведет себя в растворе как единое целое. Эта частица вместе со свободными противоионами диффузного слоя, которые сообщают заряд дисперсионной среде, образуют мицеллу
– структурную единицу коллоидного раствора.
Суммарный заряд мицеллы в силу электронейтральности равен нулю.
Знак ξ – потенциала коллоидной частицы зависит от состава дисперсионной среды в момент образования дисперсной фазы системы. Например, если система получена методом конденсации при медленном вливании разбавленного раствора нитрата серебра в более концентрированный раствор йодида калия в соответствии со следующей реакцией обмена: AgNO3
(разб) + KCl(конц) → AgCl(крист) + KNO3
, то на ядре, состоящем из кристаллов йодистого серебра, адсорбируются йодид-ионы, входящие в состав образующихся кристаллов йодистого серебра и присутствующие в системе в момент образования этих кристаллов в избытке
. Йодид-ионы будут в данном случае потенциалопределяющими (зарядообразующими) и ξ – потенциал будет отрицательным. Если же порядок смешения растворов сменить на противоположный, то в момент образования кристаллов йодистого серебра в растворе в избытке будут находиться ионы серебра, входящие в состав кристаллов. Они и будут потенциалопределяющими, и в этом случае ξ–потенциал окажется положительным. Следовательно, химический состав мицеллы зависит от многих факторов и записывается полуколичественно, что позволяет составить общее представление о свойствах коллоидной системы. В формуле указывают качественный химический состав ядра, состав коллоидной частицы с потенциалопределяющими ионами и адсорбционным слоем противоионов и состав диффузного слоя мицеллы.
Например, положительно заряженный коллоидный раствор (золь) йодида серебра имеет мицеллу следующего состава: {[(AgJ)m
∙nAg+
∙xNO3
-
](п-х)+
∙n-x)NO3
-
}
Отрицательно заряженный золь йодида серебра имеет мицеллу вида: {[(AgJ)m
∙nJ-
∙xK+
] (п-х)-
∙(n-x)K+
}
где m – число молекул йодида серебра в ядре; n и x - количество потенциалопределяющих ионов и противоионов в адсорбционном слое частицы; (n-x) – число противоионов в диффузном слое мицеллы. На Рис. 3.1. показано схематическое строение мицеллы йодида серебра с положительно заряженной коллоидной частицей.
При добавлении раствора сульфата алюминия к раствору хлорида бария можно получить золь сульфата бария (нерастворимой соли) с положительно заряженной коллоидной частицей, так как в растворе имеется избыток катионов бария. Строение такой мицеллы сульфата бария можно выразить схемой:
{[(BaSO4
)m
∙nBa2+
∙2(n-x)Cl-
] 2x+
∙2xCl-
}
3.3. Свойства коллоидных систем
Коллоидные системы можно разделить на лиофобные и лиофильные. Лиофобные коллоидные системы
необратимо взаимодействуют с дисперсионной средой, что приводит к их коагуляции. Такие системы очень чувствительны к коагулирующему воздействию электролитов. Лиофильные коллоидные системы
нечувствительны к электролитам, так как дисперсная фаза таких систем обратимо взаимодействует с дисперсионной средой, что приводит к пептизации системы.
К важнейшим свойствам коллоидных систем относится их опалесценция
(свечение, или эффект Тиндаля
), вызванный дифракцией световых лучей на коллоидных частицах. Это свойство коллоидных растворов позволяет отличать их от истинных растворов. Если поместить дисперсную систему в электрическое поле, наблюдается явление, известное под названием электрофореза
. Частицы дисперсной фазы (глина) и дисперсионной среды (вода) при наличии разности потенциалов движутся в противоположных направлениях. Электрофорезом называется перемещение в электрическом поле частиц дисперсной фазы; перемещение дисперсионной среды называется электроосмосом
. Электрофорез используют для обезвоживания нефти, в производственных процессах (воздушные электрофильтры, покрытия), в медицинской практике для введения в организм коллоидных лекарственных препаратов и т.д. Электроосмос используется при опреснении воды (обратный осмос), при электроосушении и очистке лекарственных препаратов, для дубления кожи и пр.
При объяснении свойств коллоидных систем необходимо учитывать наличие в них поверхности раздела фаз
, на которых могут происходить различные явления. На границе раздела жидкости и твѐрдого тела (а также жидкости и еѐ насыщенного пара или двух несмешивающихся жидкостей) в результате различного молекулярного взаимодействия в соприкасающихся фазах, в поверхностном слое жидкости появляется поверхностное натяжение. Поверхностное натяжение δ зависит от свободной поверхностной энергии F и площади поверхностного слоя S:
 , (3.1) , (3.1)
  где F
- изменение свободной поверхностной энергии, а S
- где F
- изменение свободной поверхностной энергии, а S
-
изменение площади поверхностного слоя.
Снижение поверхностного натяжения достигается введением в жидкость поверхностно-активных веществ (ПАВ) - мыл или моющих средств, которые адсорбируются
на поверхностях раздела и уменьшают свободную поверхностную энергию. Молекулы большинства ПАВ обладают дифильным строением, т.е. содержат как полярную группу, так и неполярный углеводородный радикал.
Расположение таких молекул в поверхностном слое энергетически наиболее выгодно при условии ориентации молекул гидрофильной полярной группой к полярной фазе (полярной жидкости), а гидрофобной неполярной группой– к неполярной фазе (газу или неполярной жидкости). Поэтому соединения с гидрофобной поверхностью, плохо смачиваемые водой, концентрируются в виде плѐнки на границе раздела фаз. Это явление называется флотацией
и используется, например, для извлечения металлов из руд. При малой концентрации раствора тепловое движение нарушает ориентацию молекул ПАВ; при повышении концентрации происходит насыщение адсорбционного слоя и на поверхности раздела фаз образуется слой вертикально ориентированных молекул ПАВ. Образование такого мономолекулярного слоя соответствует минимальной величине поверхностного натяжения
раствора ПАВ и максимальному значению адсорбции
; при дальнейшем увеличении концентрации ПАВ в растворе поверхностное натяжение и адсорбция не изменяются.
На границе раздела двух фаз может иметь место явление сорбции. Сорбция
– это концентрирование или сгущение одного вещества в другом веществе, или поглощение одного вещества (например, газа) другим (например, жидкостью или твѐрдым телом). Адсорбцией
называется поглощение вещества поверхностным слоем
жидкости или твѐрдого тела. Абсорбция
подразумевает распределение поглощаемого вещества по всему объѐму поглощающего вещества. Поглощаемое вещество называется адсорбатом
, а поглощающее - адсорбентом
. Десорбция – это процесс, обратный адсорбции, то есть отделение адсорбата от адсорбента. Адсорбированные частицы удерживаются адсорбентом некоторое время, которое зависит от природы адсорбента и адсорбата, температуры и давления. Со временем интенсивность адсорбции замедляется, а скорость процесса десорбции возрастает. Адсорбционным равновесием называется такое состояние системы «адсорбент-адсорбат», при котором скорости процессов адсорбции и десорбции сравниваются. При физической адсорбции
адсорбционные силы имеют ту же природу, что и силы межмолекулярного взаимодействия в газах, жидкостях и твѐрдых телах. При химической адсорбции
(хемосрбции
) молекула адсорбата образуют химическое соединение с адсорбентом. Физическая сорбция, в отличие от химической, обладает невысоким тепловым эффектом и обратимостью.
Адсорбция сопровождается понижением свободной энергии поверхностного слоя адсорбента. Адсорбат должен обладать меньшим, чем адсорбент, поверхностным натяжением. Количественной характеристикой адсорбции является величина адсорбции Г, определяемая как избыток адсорбата в молях на единицу площади (в см2
) поверхностного слоя по сравнению с содержанием его в таком же объѐме соприкасающихся фаз.
По уравнению Гиббса:
Г
 , (3.2) , (3.2)
где С - концентрация адсорбата в среде, из которой происходит поглощение,
моль;  - поверхностная активность адсорбата; Т – температура; R – универсальная газовая постоянная, равная 8,31 Дж/моль∙К. - поверхностная активность адсорбата; Т – температура; R – универсальная газовая постоянная, равная 8,31 Дж/моль∙К.
3.4. Устойчивость коллоидных систем
Коллоидные системы потому и рассматриваются особо, отлично от обычных гетерогенных систем, что по внешнему виду они напоминают растворы. В то же время само существование коллоидных систем противоречит законам термодинамики, так как они обладают огромным запасом поверхностной энергии, но, несмотря на это, освобождаются от нее «нехотя». Устойчивость коллоидных систем объясняется наличием у коллоидных частиц электрического заряда (ξ–потенциала). Частицы дисперсной фазы адсорбционного и диффузного слоя имеют одноименный заряд, вызывающий их взаимное отталкивание. Разность потенциалов E
между твѐрдой фазой и раствором можно определить из следующего соотношения:
 RT
(3.1) E
lnC zF RT
(3.1) E
lnC zF
где R – универсальная газовая постоянная, равная 8,31 Дж/моль∙К; T – температура в градусах Кельвина; z – заряд потенциалопределяющего иона;
F – число Фарадея, или 96500 Кл; С – концентрация потенциалопределяющего иона.
Наличие электрического заряда у коллоидных частиц приводит к гидратации, так как полярные молекулы воды вступают во взаимодействие с коллоидными частицами. Ядра коллоидных частиц многих золей имеют собственную гидратную оболочку. Такие золи называются гидрофильными
. Золи, ядра коллоидных частиц которых не взаимодействуют с водой, называются гидрофобными
. Гидратная оболочка снижает поверхностную энергию дисперсной фазы и приводит к разобщению частиц в коллоидном растворе. Это явление называется коллоидной устойчивостью. Коллоидная устойчивость
определяет способность коллоидных систем сохранять такие свойства как окраска, прозрачность, однородность. При длительном хранении гидрофильные золи переходят в особое «студнеобразное» коллоидное состояние. В таком виде их называют гелями
. Структура геля такова, что мицеллы не разрушаются, а связываются друг с другом, образуя ячейки, внутри которых сохраняется водная среда. Гель можно высушить, превратив его в твердый коллоид. Примером такого геля является желатин.
Устойчивость золя можно нарушить, устранив одноименный заряд и защитную гидратную оболочку. Коллоидные частицы в этом случае укрупняются, слипаются. Процесс укрупнения частиц, потеря агрегативной устойчивости золя называется его коагуляцией
. Процесс, обратный коагуляции, называется пептизацией
. Коагуляция золя приводит к потере его кинетической устойчивости, которая выражается в образовании осадка. Этот процесс называется седиментацией
. Одним из методов разрушения коллоидной системы – введение в нее твердого электролита или его концентрированного раствора. Ионы электролита проникают внутрь коллоидной частицы, снижая ее заряд. Это приводит к коагуляции и седиментации коллоидного раствора. Коагуляцию золя вызывает тот из ионов прибавленного электролита, заряд которого противоположен заряду коллоидной частицы. Например, коагуляцию золя сульфата бария с
положительно заряженной коллоидной частицей могут вызвать отрицательно заряженные анионы добавленного к золю электролита, а коагуляцию золя того же малорастворимого соединения , но с отрицательно заряженной коллоидной частицей, вызовут положительно заряженные катионы.
По правилу Шульце-Гарди
, коагулирующая способность иона определяется его зарядом: чем больше заряд иона, тем больше его коагулирующая способность. Так, например, в ряду анионов SO4
2-
, PO4
3-
и Cl-
наилучшим коагулирующим действием для золя йодида серебра с положительно заряженной коллоидной частицей будет обладать фосфат-анион PO4
3
, обладающий наибольшим отрицательным зарядом.
Минимальное количество электролита, прибавленного к золю, которое может вызвать его коагуляцию, называется порогом коагуляции электролита
. Он измеряется в миллимоль10
/л и рассчитывается по следующему соотношению:
10
1 миллимоль равен 10-3
моль
 NV
1
1000 (3.3) NV
1
1000 (3.3)
ПК V
1 V
2
где N - нормальная концентрация электролита; V1
– объем электролита, л; V2
– объем золя, л
Значение порогов коагуляции электролитов с одно-, двух- и трехзарядными ионами относятся как числа 29:11:1.
Коагуляция золя может быть вызвана его взаимодействием с другим золем, частицы которого имеют противоположный заряд. Так, смешение золя гидроксида железа
({[Fe(OH)3
]m
∙nFe3+
∙3(n-x)Cl–
}3x+
∙3xCl-
),
коллоидные частицы которого имеют положительный заряд, с отрицательно заряженным золем сульфида мышьяка
({[Аs2
S3
]m
∙nS2–
∙2(n-x)Н+
}2x–
∙2xН+
)
приводит к их взаимной коагуляции. В данном случае коагуляция обусловлена тем, что коллоидные частицы одного вида являются как бы очень крупными многозарядными ионами – коагулянтами для частиц другого вида. Взаимная коагуляция коллоидных систем может наблюдаться и тогда, когда частицы золей имеют одноименный заряд; в этом случае причиной потери устойчивости одного из золей является адсорбция потенциалообразующего иона одной системы поверхностью коллоидных частиц другой системы.
3.5. Вопросы и задания:
1. Чем дисперсные системы отличаются от растворов?
2. К какой дисперсной системе относится молоко? Пенопласт? Мыльная пена? Дым? Жемчуг?
3. Как устроена мицелла? Что такое дзета-потенциал?
4. Какой ион является потенциалопределяющим для золя гидроксида железа Fe(ОН)3
, образующегося при гидролизе (реакции с водой) хлорида железа FeCl3
?
5. Золь кремниевой кислоты H2
SiO3
был получен при взаимодействии растворов K2
SiO3
и HCl. Напишите формулу мицеллы золя и определите, какой из электролитов был в избытке, если в процессе электрофореза противоионы движутся в электрическом поле к катоду.
6. При пропускании избытка сероводорода в раствор соли мышьяка (III) AsCl3
получили золь сульфида мышьяка. Учитывая условия образования, напишите формулу мицеллы золя и определите знак его заряда (ξ–потенциала).
7. Чем физическая адсорбция отличается от химической?
8. Как изменяется величина поверхностного натяжения при растворении в воде поверхностно-активного вещества?
9. Как расположатся пороги коагуляции в ряду электролитов CrCl3
, Ba(NO3
)2
, K2
SO4
для золя кремниевой кислоты, частицы которого заряжены отрицательно?
10. В каждую из трех колбу налито по 0,01 л золя хлорида серебра. Для коагуляции в 1-ю колбу добавлено 0,002л 0,1н раствора нитрата натрия, во 2-ю – 0,012л 0,01н раствора нитрата кальция, а в 3-ю – 0,007л 0,001н раствора нитрата алюминия. Вычислите пороги коагуляции электролитов, определите знак заряда золя. (Ответ: 1- 166,7 ммоль/л; 2 – 5,45 ммоль/л; 3 – 0,41 ммол/л)
Лекция 4. Электрохимические системы
В электрохимических системах происходят процессы взаимного превращения химической и электрической энергии, которые называются электрохимическими процессами
. Электрохимические процессы можно разделить на две группы: 1) процессы превращения химической энергии в электрическую (в гальванических элементах);
и 2) процессы превращения электрической энергии в химическую (электролиз)
.
4.1. Гальванические элементы
Гальванический элемент- это электрохимическая система, состоящая из двух электродов и одного или нескольких растворов электролитов, в которой происходит превращение химической энергии в электрическую Простейшая такая система – это два электрода, каждый из которых опущен в раствор собственной соли. Рассмотрим процессы, протекающие при погружении металла в раствор его соли. Металлы имеют кристаллическое строение. Ионы в узлах кристаллической решетки металла находятся в равновесии со свободными электронами. Ме ↔ Меn+
+ nе-
(4.1)
При погружении металла в раствор начинается сложное взаимодействие металла с компонентами раствора. Наиболее важным является взаимодействие поверхностных ионов металла с полярными молекулами воды, смещающее равновесие. В результате ионы металла переходят в раствор:
Ме → Меn+
+ ne-
(4.2)
При этом металл становится заряженным отрицательно, а раствор положительно. На границе металл-раствор возникает двойной электрический слой
. Между металлом и раствором возникает разность потенциалов, которая называется электродным потенциалом,
или потенциалом электрода. Наряду с реакцией (4.2) протекает обратная реакция:
Меn+
+ ne-
→ Me (4.3)
При некотором значении электродного потенциала скорость прямого процесса будет равна скорости обратного процесса; устанавливается новое равновесие, а потенциал, соответствующий условиям равновесия, называется равновесным электродным потенциалом
.
Рассмотрим систему, которая состоит из медной пластинки, опущенной в раствор сульфата меди, и цинковой пластинки, опущенной в раствор сульфата цинка. На поверхности цинковой пластинки возникает двойной электрический слой и устанавливается равновесный электродный потенциал в результате процесса:
Zn = Zn+2
+ 2e-
.
На поверхности медной пластинки происходят такие же процессы и в результате реакции
Cu = Cu+2
+ 2e-
на поверхности медной пластины также возникает равновесный электродный потенциал.
Электродные потенциалы цинка и меди различны по величине, поэтому если соединить медную и цинковую пластинки проводником, а растворы – солевым мостиком, то электроны будут переходить от цинка к меди. Равновесие на цинковом электроде сместится влево, и в раствор будет переходить больше ионов цинка (цинк будет растворяться), а на медном электроде равновесие сместится влево, и ионы меди из раствора будут разряжаться (получать электроны) на медной пластине (медь будет осаждаться). Эти процессы будут продолжаться до тех пор, пока не растворится весь цинк, или не высадится на медном электроде вся медь. При этом в проводнике будет иметь место переход электронов от цинка к меди, то есть по цепи потечѐт электрический ток
.
Рассмотренная схема называется гальваническим элементом Даниэля-Якоби
, при работе которого протекают следующие процессы:
1) Реакция окисления цинка. Процессы окисления в электрохимии называются анодными процессами,
а электроды, на которых идут процессы окисления, называются анодами
.
2) Реакция восстановления ионов меди. Процессы восстановления в электрохимии называются катодными процессами,
а электроды, на которых идут процессы восстановления, называются катодами
.
3) Движение электронов во внешней цепи;
4) Движение ионов в растворе: анионов SO4
-2
к аноду, а катионов Cu+2
и Zn+2
к катоду.
Вследствие химической реакции
Zn + Cu+2
= Cu + Zn+2
(4.4)
в гальваническом элементе возникает электрический ток
(движение электронов во внешней цепи и движение ионов в растворе).
Приведенная выше реакция (4.4.) называется токообразующей
.
При схематической записи гальванического элемента границу раздела между проводником первого рода
(металлом) и проводником второго рода
(раствором) обозначают одной чертой, а между проводниками второго рода – двумя чертами. Схема гальванического элемента Даниэля-Якоби имеет вид:
(-) анод Zn/Zn+2
//Cu+2
/Cu катод (+)
С помощью гальванического элемента можно совершить электрическую работу
за счет энергии химической реакции. Электрическая работа равна произведению разности потенциалов на количество электричества. Максимальная разность потенциалов электродов, которая может быть получена при работе гальванического элемента, называется электродвижущей силой
(ЭДС) элемента Е. Она равна разности равновесных потенциалов катода φ1
и анода φ2
:
E
 (4.5) (4.5)
Абсолютные значения электродных потенциалов экспериментально определить невозможно. Однако можно определить разность электродных потенциалов измеряемого электрода и электрода, потенциал которого условно принимают равным нулю
. В настоящее время равным нулю считается потенциал стандартного водородного электрода.
Такой электрод состоит из платинированной платины (на поверхность платины наносят слой высокодисперсной платиновой черни), контактирующей с газообразным водородом, находящимся под давлением 101кПА (1 атмосфера), и раствором, в котором активность ионов водорода равна 1 моль/л. Платина непосредственно в реакции не участвует. Водород адсорбируется (поглощается) на платине, взаимодействует с молекулами воды и переходит в раствор в виде ионов, оставляя на платине электроны. При этом платина заряжается отрицательно, а раствор положительно, то есть возникает электродный потенциал. Фактическая величина его неизвестна, и ее условно принимают равной нулю
. Равновесие на водородном электроде можно представить реакцией: 2Н+
+ 2е-
↔ Н2
Для определения потенциалов электродов собирают гальванический элемент, состоящий из водородного электрода и измеряемого электродов. На Рис. 4.1. показана схема измерения электродного потенциала посредством водородного электрода. Металл, потенциал которого измеряется, может служить по отношению к водородному электроду либо катодом, либо анодом. Тогда, в соответствии с (4.5), будет меняться знак измеряемого потенциала.
Рис. 4.1. Измерение потенциала металлического электрода (слева) по водородному электроду (справа). Концентрация ионов водорода [Н+]
в растворе равна 1 М, давление Н2
– 1 атмосфера.
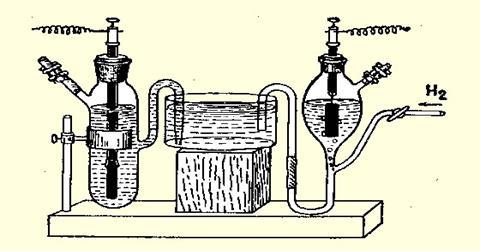
Схема гальванического элемента для измерения потенциала, к примеру, цинкового электрода имеет вид:
Н2
, Pt / H+
// Zn+2
/Zn
Токообразующей реакцией в водородно-цинковом элементе будет следующая реакция:
Zn + 2H+
= Zn+2
+ H2
Разность потенциалов катода и анода
Е = φ
Zn
- φH2
,
то есть ЭДС элемента, будет равна потенциалу цинка. Потенциал цинка имеет знак «-«, так как в данном гальваническом элементе цинк является анодом. Если измерить потенциал меди, то она, как менее активный металл, будет катодом. Разность потенциалов катода и анода будет равна Е = φ
н2
– φcu,
то есть потенциалу меди, который имеет положительный заряд. Обычно потенциалы обозначаются как φ
(или Е
) с индексами исходных веществ и продуктов реакции, например,
φ
Zn+2/Zn или φ
Cu+2/Cu.
Практически при измерениях потенциалов пользуются не водородным электродом, а более удобными в обращении электродами, потенциалы которых по отношению к водородному электроду известны.
Это хлорсеребряный и каломельный электроды.
Стандартным потенциалом (φo
) металлического электрода называют потенциал этого электрода в растворе его собственных ионов, активность (концентрация) которых равна единице. Такая концентрация называется стандартной.
Потенциал электрода, когда концентрации ионов в растворе отличны от стандартной, определяется по уравнению Нернста:
 lga
(4.6) n lga
(4.6) n
где φ 0
– стандартный электродный потенциал; n – число электронов, участвующих в электродном процессе; а – активность катионов металла в растворе. В разбавленных растворах активность можно заменить концентрацией ионов [C] в моль/л.
Если составить гальванический элемент из одинаковых электродов, то ЭДС такого элемента будет определяться разностью активностей (концентраций) ионов металла в растворах электролитов. Такие гальванические элементы называются концентрационными
.
Пример: Найти ЭДС гальванического элемента, составленного по следующей схеме: Анод (-) Fe/0,01M FeSO4
//0,1M FeSO4
/Fe Катод (+).
Решение: По уравнению Нернста (4.6)
 E E
 0,028в 0,028в
Таблицу с занесенными в неѐ значениями стандартных электродных потенциалов называют рядом стандартных электродных потенциалов, или рядом напряжений, или рядом активности металлов. В Таблице 4 ниже приведены значения электродных потенциалов распространѐнных металлов.
Таблица 4. 1. Стандартные электродные потенциалы
| Электрод
|
Eо
, В
|
Электрод
|
Eо
, В
|
Электрод
|
Eо
, В
|
| Литий
|
-3,045
|
Марганец
|
-1,18
|
Свинец
|
-0,126
|
| Калий
|
-2,924
|
Хром
|
-0,913
|
Водород
|
0,000
|
| Барий
|
-2,90
|
Цинк
|
-0,763
|
Медь
|
+0,34
|
| Кальций
|
-2,87
|
Железо
|
-0,44
|
Ртуть
|
+0,79
|
| Натрий
|
-2,714
|
Кадмий
|
-0,403
|
Серебро
|
+0,80
|
| Магний
|
-2,37
|
Никель
|
-0,25
|
Платина
|
+1,19
|
| Алюминий
|
-1,70
|
Олово
|
-0,136
|
Золото
|
+1,50
|
Стандартные электродные потенциалы указывают на меру восстановительной способности атомов металла, а также на меру окислительной способности его ионов. Чем более отрицательное значение имеет потенциал металла, тем более сильной восстановительной способностью обладает металл. Чем больше разность потенциалов металлов в ряду стандартных электродных потенциалов, тем большую ЭДС в гальванических элементах они могли бы дать. Более активный металл
служит в гальваническом элементе анодом
.
В электрохимических системах критерием возможности самопроизвольного протекания электрохимической реакции является отрицательное значение изменения энергии Гиббса
 G (см. Лекцию 13). G (см. Лекцию 13).
 G nFE
(см. 13.9) G nFE
(см. 13.9)
где n – число электронов, участвующих в токообразующей реакции; F – число Фарадея, а E – электродвижущая сила (ЭДС), или напряжение источника тока в отсутствие внешней нагрузки.
Если внешние условия отличаются от стандартных, потенциал электрода меняется и отличается от стандартного. Это явление называется поляризацией электрода
. Концентрационной поляризацией
называется изменение потенциала электрода вследствие изменения концентраций реагентов в приэлектродном слое при прохождении тока. Концентрационная поляризация определяется по уравнению Нернста в следующем виде:
 поляр
0,059 C
1
(4.7) поляр
0,059 C
1
(4.7)
E
lg
n C
2
где с1
и с2
соответственно меньшая и большая концентрации иона, моль/л
Перенапряжением
 называют повышение потенциала разрядки ионов по сравнению со значением стандартного потенциала в равновесных условиях вследствие замедленности собственно электрохимических стадий реакции. Значения перенапряжения для выделения водорода и кислорода на некоторых металлах приводятся в соответствующих таблицах. называют повышение потенциала разрядки ионов по сравнению со значением стандартного потенциала в равновесных условиях вследствие замедленности собственно электрохимических стадий реакции. Значения перенапряжения для выделения водорода и кислорода на некоторых металлах приводятся в соответствующих таблицах.
Пример: Как изменится ЭДС при работе гальванического элемента со схемой: (-)2Al|2Al3+|
HCl10-4
M|(Pt)(3H2
|6H+
(+), если в процессе работы
концентрации ионов алюминия меняется от 0,003М до 0,1М, а перенапряжение водорода на платине равно 0,09В? Чему равна концентрационная поляризация анода?
Решение: Определяем по (4.6) первоначальные электродные потенциалы катода и анода. Начальный потенциал алюминиевого
 электрода равен: lg3 10 3
1,71В
.
Начальный электрода равен: lg3 10 3
1,71В
.
Начальный
 / Al
/ Al / Al
/ Al
потенциал водородного электрода равен:
 lg100,23В
.
Алюминиевый электрод является lg100,23В
.
Алюминиевый электрод является
 / H
2
/ H
2 / H
2
/ H
2
анодом, а водородный катодом, поэтому начальная ЭДС ГА равна: -0,23 – (-1,71) = 1,48В. Концентрационную поляризацию анода (алюминиевого
 поляр
0,059 C
2
0,059 lg 0,1 0,029В
электрода) находим по (4.7): E
lg поляр
0,059 C
2
0,059 lg 0,1 0,029В
электрода) находим по (4.7): E
lg
n C
1
3 0,003
   Потенциал алюминиевого электрода с учѐтом поляризации равен: а
1,71 0,029 1,68В
. Потенциал водородного электрода с учѐтом перенапряжения водорода на платине равен: к
0,23 0,09 0,32В
. Конечная ЭДС ГА равна: к а
0,32 ( 1,68) 1,36В
. Потенциал алюминиевого электрода с учѐтом поляризации равен: а
1,71 0,029 1,68В
. Потенциал водородного электрода с учѐтом перенапряжения водорода на платине равен: к
0,23 0,09 0,32В
. Конечная ЭДС ГА равна: к а
0,32 ( 1,68) 1,36В
.
Число реакций, используемых в химических источниках электрической энергии, невелико. Это связано с тем, что не всякий гальванический элемент обладает ценными техническими свойствами (высокая и постоянная ЭДС, возможность отбирания больших токов, длительность работы и сохранность) и относительной дешевизной. Почти во всех выпускаемых в настоящее время гальванических элементах анод изготавливается из цинка, а в качестве катода используют оксиды менее активных металлов. Химические источники электрической энергии применяются в различных областях промышленности. Однако существующие в настоящее время гальванические элементы довольно дороги. Так, стоимость электроэнергии, вырабатываемой обычной батарейкой для карманного фонарика почти в 800 раз выше стоимости электроэнергии, поставляемой промышленными электростанциями. Однако, гальванические элементы незаменимы, когда существует необходимость в портативных источниках электроэнергии.
Все химические источники тока делятся на необратимые (гальванические элементы) и обратимые (аккумуляторы)
. Первые используются однократно, вторые предназначены для многократного использования. Примером химического источника тока первой группы может служить гальванический элемент Даниэля-Якоби.
В качестве примера обратимого источника тока рассмотрим действие свинцового аккумулятора. Свинцовый аккумулятор представляет собой электрохимическую систему PbO2
/H2
SO4
/Pb с ЭДС, равной 2,1 В. Электроды аккумулятора состоят из свинцовых ячеек, заполненных пастой из оксида свинца. В качестве электролита используют 30%-й раствор серной кислоты. При погружении электродов в кислоту на поверхности пластин образуется малорастворимый сульфат свинца. При зарядке аккумулятора происходят следующие процессы:
Катодный процесс (на отрицательном электроде):
PbSO4
+ 2H+
+ 2e = Pbтв + H2
SO4
Анодный процесс (на положительном электроде):
PbSO4
+ 2H2
O –2e = PbO + H2
SO4
+ 2H+
При работе аккумулятора (разрядке) на аноде идет окисление:
(А) Pb + H2
SO4
= PbSO4
+ 2H+
+ 2e
На катоде идет восстановление:
(К) PbO + H2
SO4
+2H+
= PbSO4
+ 2H2
O –2e
Самым современным и дешевым является топливный гальванический элемент
. Топливным называется гальванический элемент, в котором электрохимически активными веществами служат традиционное топливо и кислород, а генерирование электрической энергии происходит за счет окисления топлива
. Сгорание топлива в топливном элементе происходит непосредственно на его электродах, что исключает неполное сгорание, потери энергии и «тепловое засорение» окружающей среды. Топливный элемент обладает высоким КПД. На данном этапе развития его удалось довести до 70%, а можно довести до 80% и более. Полые электроды топливного элемента изготовлены из пористого угля, пропитанного катализатором. В полость катода поступает топливо (в идеале водород), а в полость анода поступает кислород. Электролитом служит 30-40% водный раствор гидроксида калия. Во время работы элемента топливо проникает через поры катода на его наружную поверхность, где окисляется по схеме:
Н2(
г) + 4ОН-
(р) – 4е = 4Н2
О(ж)
Одновременно с этим кислород проникает через поры анода на его наружную поверхность, где восстанавливается по схеме: О2
(г) + 2Н2
О(ж) + 4е = 4ОН-
Суммарная токообразующая реакция имеет вид:
2Н2
(г) + О2
(г) = 2Н2
О(ж).
Если в топливном элементе используется не водород, а другое топливо, то реакции на электродах будут иные.
Топливные элементы компактнее других гальванических элементов, поэтому их успешно используют на космических кораблях, подводных лодках и т.п. В последнее время ведутся исследования, направленные на изготовление неприхотливых гальванических элементов, работающих на нефтяном топливе и даже каменном угле. Большой интерес представляет собой возможность хранения водорода в связанном состоянии в виде легко разлагающихся гидридов металлов (лития, титана и др.).
4.2. Электролиз
Электролизом называют совокупность процессов, имеющих место при прохождении постоянного электрического тока через электрохимическую систему, состоящую из двух электродов и расплава или раствора электролита.
Простейшим электрохимическим процессом такого рода является электролиз расплавов. Рассмотрим, например, электролиз расплава хлористого магния. При прохождении через расплав тока катиона магния под действием электрического поля движутся к отрицательному электроду. Здесь, взаимодействуя с приходящими по внешней цепи электронами, они восстанавливаются по схеме: Mg2+
+ 2e → Mg
Анионы хлора перемещаются к положительному электроду и, отдавая избыточные электроны, окисляются:
2Cl-
-2e → Cl2
Складывая уравнения процессов, протекающих у электродов, получаем суммарное уравнение окислительно-восстановительной реакции, происходящей при электролизе расплава хлористого магния: Mg2+
+ 2Cl-
→ Mg + Cl2
Эта реакция не может протекать самопроизвольно, энергия, необходимая для ее осуществления, поступает от внешнего источника постоянного тока.
Электрод, на котором происходит окисление, называется анодом; электрод, на котором происходит восстановление, называется катодом. Однако, в отличие от гальванического элемента, при электролизе катод заряжен отрицательно, а анод – положительно, то есть распределение знаков заряда электродов противоположно тому, которое имеет место при работе гальванического элемента
. Следовательно, на отрицательном электроде (катоде) происходит катодное восстановление ионов магния до металлического магния, а на положительном электроде (аноде) идет анодное окисление ионов хлора до газообразного хлора.
При электролизе водных растворов электролитов необходимо иметь в виду, что кроме ионов электролита, в растворе находятся ионы водорода Н+
и гидроксила ОН-
,
являющиеся продуктами диссоциации воды. В электрическом поле ионы водорода перемещаются к катоду, а ионы гидроксила к аноду. Таким образом, у катода могут разряжаться как катионы электролита, так и катионы водорода. Аналогично, у анода может происходить разряд как анионов электролита, так и анионов гидроксила. Молекулы воды могут также подвергаться электрохимическому восстановлению или окислению. Из нескольких возможных процессов будет протекать тот, осуществление которого сопряжено с минимальной затратой энергии.
Рассматривая катодные процессы, необходимо учитывать потенциал восстановления ионов водорода. Этот потенциал зависит от концентрации ионов водорода и в случае нейтральных растворов (при рН = 7) равен –0,41в. Поэтому, если катионом электролита является металл, электродный потенциал которого значительно положительнее, чем –0,41в
, то будет происходить восстановление иона металла. Такие металлы находятся после олова включительно. Наоборот, если катионом является металл, имеющий потенциал значительно более отрицательный, чем –0,41в, то металл восстанавливаться не будет, а будет происходить выделение водорода по схеме: 2Н+
+ 2е = Н2
К таким металлам относятся металлы начала ряда напряжений, приблизительно до титана. Наконец, если потенциал металла близок к значению 0,41в (это металлы средней части ряда напряжений – цинк, хром, железо, кадмий, никель), то в зависимости от условий электролиза возможно как восстановление металла, так и выделение водорода; нередко наблюдается одновременно и то, и другое.
Кислородсодержащие анионы разряжаются труднее, чем анион гидроксила. Поэтому на аноде будет происходить окисление ионов гидроксила по схеме:
4ОН-
- 4е = О2
+ 2Н2
О
в щелочной среде
2Н2
О – 4е = О2
+ 4Н+
в кислой или нейтральной среде.
. При электролизе водных растворов бескислородных кислот и их солей у анода разряжаются их анионы (кроме плавиковой кислоты и фторидов).
Например, при электролизе водного раствора хлористого натрия на катоде выделяется водород, а на аноде хлор:
Катод (-) NaCl + Н2
О Анод (+)
Na+
(-2,7в) 2Cl-
(+1,36в) Н+
(-0,41в) ОН-
(+1,22в) 2Н+
+ 2е
→ Н2
Na+
и OH-
→NaOH
2Сl-
-2e
→ Cl2
Суммарный процесс, идущий в данной электрохимической системе:
NaCl + Н2
О → Н2
+ Cl2
+ NaOH
При электролизе раствора сернокислой меди на катоде происходит выделение меди, а на аноде выделение кислорода:
| Катод (-)
|
CuSO4
+ Н2
О
|
Анод (+)
|
| |
Сu2+
(+0,337в) SO4
2-
(+2,01в)
|
|
| |
Н+
(-0,41в) ОН-
(+1,22в)
|
|
| Cu2+
+2e
→ Cu
|
2Н
+ + SO
42-
→ H2SO4 CuSO4
+H2
O→Cu+O2
+H2
SO4
|
4ОН-
- 4е
→ О2
+
2Н2
О
|
При электролизе раствора сернокислого натрия на катоде выделяется водород, а на аноде кислород.
| Катод (-)
|
Na2
SO4
+ Н2
О
|
Анод (+)
|
| |
Na+
(-2,714в) SO4
2-
(+2,01в)
|
|
| |
Н+
(-0,41в) ОН-
(+1,22в)
|
|
| 2Н+
+ 2е
→ Н2
|
2Na
+ + SO
42-
→ Na
2SO
4
H2
O
→H2
+O2
|
4ОН-
- 4е
→ О2
+
2Н2
О
|
В данном случае фактически происходит электролитическое разложение воды, а сернокислый натрий просто увеличивает электропроводность раствора.
Равновесие в процессах электролиза смещается вправо, поскольку в систему поступают электроны в виде постоянного тока.
Характер катодного процесса, как мы уже видели, определяется, прежде всего, положением соответствующего металла в ряду напряжений. При рассмотрении анодных процессов следует учитывать, из какого материала изготовлен анод. Различают электролиз с инертным анодом и электролиз с активным анодом
. Инертным называется анод, материал которого не претерпевает окисления в ходе электролиза. В качестве материала для инертных анодов чаще всего применяют графит, уголь или платину. Активный анод окисляется в процессе электролиза. В случае активного анода число конкурирующих анодных процессов возрастает до трех, а именно:
1. Электрохимическое окисление воды с выделением кислорода; 2. Разряд аниона
3. Электрохимическое окисление металла анода (так называемое анодное растворение металла).
Из всех этих возможных процессов будет идти тот, который энергетически наиболее выгоден. Если потенциал металла анода располагается в ряду стандартных потенциалов раньше обеих других электрохимических систем, то будет наблюдаться анодное растворение металла. В противном случае будет происходить выделение кислорода или разряд аниона. Рассмотрим, например, электролиз раствора никеля с никелевым электродом. На катоде происходит, в основном, разряд ионов никеля и выделение металла, так как потенциал никеля (- 0,250 в) значительно больше –0,41в. На аноде происходит окисление никеля, так как потенциал никеля намного меньше потенциала окисления воды (+1,23в), а тем более потенциала окисления сульфат-иона (+2,01в):
Катод (-) NiSO4
+ Н2
О Анод (+) Ni Ni2+
(+0,250в) SO4
2-
(+2,01в)
Н+
(-0,41в) ОН-
(+1,22в) Ni2+
+2e → Ni ←Ni2+
Ni -2e → Ni2+
Таким образом, в данном случае электролиз сводится к растворению металла анода и выделению его на катоде. Этот процесс называется электролитическим рафинированием
и применяется для электролитической очистки никеля (и других металлов).
Законы электролиза были установлены выдающимся английским физиком М. Фарадеем в 30-х годах девятнадцатого века. Первый закон Фарадея гласит: Масса образующегося при электролизе вещества пропорциональна количеству прошедшего через раствор электричества.
Этот закон вытекает из сущности электролиза. В месте соприкосновения металла с раствором происходит взаимодействие ионов или молекул раствора с электронами металла, так что электролитическое образование вещества является результатом этого процесса. Поэтому количество образовавшегося вещества всегда будет пропорционально числу прошедших по цепи электронов, или количеству электричества.
По второму закону Фарадея, при электролизе различных химических соединений равные количества электричества приводят к электрохимическому превращению эквивалентных количеств веществ.
Например, при электролизе хлорида меди (II) каждый ион меди получает от катода два электрона, и в то же время два хлоридиона отдают аноду два электрона, превращаясь в атомы хлора. Следовательно, число выделившихся атомов меди всегда будет вдвое меньше количества выделившихся атомов хлора, то есть массы меди и хлора будут относиться друг к другу как их эквивалентные массы.
Измерениями установлено, что для превращения одного эквивалента вещества необходимо количество электричества, равное округленно 96500 кулонам, или амперам в секунду. Эту величину называют числом Фарадея и обозначают буквой F.
Оба закона Фарадея объединяются в один соотношением:
 m
ЭIt
,
(4.8) F m
ЭIt
,
(4.8) F
где Э – эквивалент выделяющегося на электроде вещества; I – сила тока в амперах; t - время в секундах, а F – число Фарадея (96500 ампер∙сек).
Во многих случаях на практике вследствие побочных процессов выделенная на электродах масса вещества Mфактическая
всегда меньше, чем рассчитанная по закону Фарадея Mтеоретическая
. Для характеристики истинного количества вещества, выделяющегося на электродах, введено понятие выхода по току Вт, которое рассчитывают по соотношению:
В
т
 100% (4.9) 100% (4.9)
т
Электролиз широко применяется в производственной и научной практике. Посредством электролиза водных растворов солей получают и очищают многие металлы. Электролитическое выделение металлов из руд называется электроэкстракцией
, а очистка металлов от примесей при помощи электролиза называется электрорафинированием
. Такое производство тяжелых и цветных металлов получило общее название гидрометаллургии
. Методом электроэкстракции получают, главным образом, цинк, медь и кадмий. Электролитическому рафинированию подвергают мель, никель, свинец, олово, серебро, золото. Электролизом расплавов в промышленности получают алюминий, магний, натрий, литий, кальций, титан и другие металлы, потенциалы выделения которых из водных растворов солей более отрицательны, чем потенциал водорода. При электролизе водных растворов щелочных металлов выделяют хлор, водород, а также получают каустическую соду (едкий натр). В результате электролиза водных растворов щелочей получают водород и кислород высокой чистоты. На электролизе водных растворов солей основано также электроосаждение
– выделение металла на катоде в виде плотного или порошкообразного осадка.
Процессы гальваностегии
представляют собой нанесение путем электролиза на поверхность металлических изделий слоев других металлов для защиты их от коррозии, придания им твердости или в декоративных целях. Это так называемые гальванические покрытия
. При нанесении гальванопокрытий катодом служит обрабатываемое изделие, а анодом – или металл покрытия, или нерастворимый электрод. На катоде происходит выделение металла покрытия. Важнейшими гальванотехническими процессами являются хромирование, цинкование и никелирование. Гальванопластикой
называют процессы получения точных металлических копий с рельефных предметов электроосаждением металла. Путем гальванопластики изготовляют матрицы для изготовления различных пластмассовых изделий (например, пуговиц), матрицы для тиснения кожи и бумаги, типографские клише, радиотехнические схемы.
К гальванотехнике относится также оксидирование, или анодирование
– покрытие поверхности металлического изделия слоем его оксида путем анодного окисления металла. Путем электросинтеза
получают пероксид водорода, перманганат калия, хроматы, хлораты и гипохлориты, а также некоторые органические вещества, например, анилин из нитробензола. Велико значение электролиза и в лабораторной практике. В основе таких методов анализа, как электровесовой, вольамперометрический и кулонометрический лежит процесс электролиза.
4.3. Коррозия металлов
Коррозией называется разрушение металлов в результате их физико-химического взаимодействия с окружающей средой.
При этом металлы окисляются и образуются продукты коррозии, состав которых зависит от условий коррозии.
Коррозия приводит к большим потерям металлов в результате разрушения трубопроводов, цистерн, металлических частей машин, корпусов судов, морских сооружений и пр. Безвозвратные потери металлов от коррозии составляют 10% от ежегодного их выпуска. По ориентировочным подсчетам, мировая потеря металла от коррозии выражается величиной 20 миллионов тонн в год. Однако, затраты на ремонт или на замену деталей судов, автомобилей, аппаратуры химических производств, приборов во много раз превышают стоимость металла, из которого они изготовлены. Таким образом, борьба с коррозией представляет собой серьѐзную экономическую проблему.
Различают химическую и электрохимическую коррозию
.
Химическая коррозия характерна для сред, не проводящих электрический ток. По условиям протекания коррозионного процесса различают: а) газовую коррозию – в газах и парах без конденсации влаги на поверхности металла, обычно при высоких температурах; б) коррозию в неэлектролитах – агрессивных органических жидкостях, таких как, например, сернистая нефть и др.
Газовая коррозия протекает по схеме: n Me + m/2O2
= Men
Om
;
Коррозию в неэлектролитах, содержащих серу, можно выразить схемой: Me + S = MeS.
Электрохимическая коррозия может протекать: а) в водных растворах электролитов, то есть солей, кислот и щелочей; б) в атмосфере любого влажного газа; в) в почве.
В воде обычно содержится растворенный кислород, способный к восстановлению по схеме:
О2
+ 4Н+
+ 4е = 2 Н2
О (1)
Кроме того, в воде присутствуют ионы водорода, также способные к восстановлению:
2Н+
+ 2е = Н2
(2)
Коррозия с участием кислорода называется коррозией с поглощением кислорода, или коррозией с кислородной деполяризацией
. Коррозия с участием ионов водорода называется коррозией с водородной деполяризацией
.
Потенциал, отвечающий электродному процессу (1), равен в нейтральной среде 0,8 в. Следовательно, растворенный в воде или нейтральных растворах кислород будет окислять те металлы, потенциал которых меньше, чем 0,8 в.
Эти металлы расположены в ряду напряжений, начиная от его начала, до серебра.
Потенциал электродного процесса (2) в нейтральной среде равен – 0,41в. Следовательно, ионы водорода в нейтральных водных растворах могут окислить только те металлы, потенциал которых меньше, чем 0,41в. Это металлы от начала ряда напряжений до кадмия.
Пример 1.
Рассмотрим электрохимическую коррозию железа в кислой среде.
На анодных участках происходит окисление железа:
(а) Fe – 2e = Fe+2
;
На катодных участках происходит восстановление водорода:
2Н+
+ 2е = Н2
.
Пример 2.
Если гвоздь вбить во влажное дерево, то коррозии подвергается (покрывается ржавчиной) та его часть, которая находится внутри дерева. Это объясняется тем, что влага древесины содержит растворенный кислород, то есть, происходит коррозия железа по схеме:
(а) Fe –2e = Fe2+
; (к) О2
+ 2Н+
+ 4е = 2Н2
О;
Продуктами коррозии являются вода и оксид железа (II), который в присутствии кислорода окисляется до оксида трехвалентного железа Fe2
O3
.
Кадмий и металлы, близкие к нему в ряду напряжений, имеют на своей поверхности защитную оксидную пленку, которая препятствует взаимодействию этих металлов с водой. Поэтому количество металлов, которые может окислить водород в нейтральной среде, еще меньше.
Таким образом, вода, содержащая растворенный кислород (в воде его обычно содержится от 0 до 14 мг/л), значительно опаснее в коррозионном отношении, чем вода, способная окислять металлы только за счет ионов водорода.
При использовании металлических материалов очень важным является вопрос о скорости их коррозии
. Кроме природы металла и окислителя и содержания последнего, на скорость коррозии могут влиять различные примеси, содержащиеся как в самом металле, так и в коррозионной среде: атмосфере или растворе. Могут иметь место различные случаи электрохимической коррозии.
Атмосферная коррозия
– это коррозия во влажном воздухе при обычных температурах. Поверхность металла, находящегося во влажном воздухе, бывает покрыта пленкой воды, содержащей различные газы и, в первую очередь, кислород. Скорость атмосферной коррозии зависит от многих факторов. В частности, на нее влияет влажность воздуха и содержание в нем газов, образующих с водою кислоты (например, СО2
или SО2
). Большое значение имеет также состояние поверхности металла: скорость атмосферной коррозии резко возрастает при наличии на поверхности шероховатостей, микрощелей, пор, зазоров и других мест, облегчающих конденсацию в них влаги.
Коррозия в грунте
(почвенная коррозия) приводит к разрушению проложенных под землей трубопроводов, оболочек кабелей, деталей строительных сооружений. Металл в этих условиях соприкасается с влагой грунта, содержащей растворенный кислород. В зависимости от состава грунтовых вод, а также минералогического состава грунта, скорость этого вида коррозии может быть различной.
Контактная коррозия
протекает, когда два металла в различными потенциалами соприкасаются друг с другом либо во влажной среде, либо при наличии влаги, конденсирующейся из воздуха. Если изделие состоит из различных металлов, то при наличии контакта между ними в присутствии растворителя изделие становится подобным работающему гальваническому элементу. Электрохимическая коррозия включает процессы анодного растворения металла и катодного восстановления окислителя. При этом металл, обладающий более отрицательным электродным потенциалом (более активный металл), окисляется (разрушается) так, словно он является анодом работающего гальванического элемента.
Пример 3.
Хром находится в контакте с медью. Какой из металлов будет окисляться, если эта пара металлов попадет в кислую среду? Составьте схему образующегося при этом гальванического элемента.
Хром более активный металл, чем медь (потенциал хрома равен – 0,744в, а меди +0,337в), поэтому в образующейся гальванической паре он будет анодом, а медь – катодом. Хромовый анод растворяется:
(а) 2Cr – 6e = 2Cr3+
;
на медном катоде выделяется водород:
(к) 6Н+
+ 6е = 3Н2
.
Схема образующегося гальванического элемента:
(-) 2Cr/Cr3+
//HCl/(Cu)3H2
/6H+
(+)
Основным отличием процессов контактной электрохимической коррозии от процессов, происходящих в гальваническом элементе, является отсутствие внешней электрической цепи.
Электроны в процессе коррозии не выходят за пределы коррозирующего металла, а двигаются внутри него. Химическая энергия преобразуется в данном случае не в электрохимическую энергию, а в тепловую
. Если изделие состоит из различных металлов, то при наличии контакта между ними в присутствии растворителя изделие становится подобным работающему гальваническому элементу. Электрохимическая коррозия включает процессы анодного растворения металла и катодного восстановления окислителя. При этом металл, обладающий меньшим электродным потенциалом (более активный металл), окисляется (разрушается) так, словно он является анодом работающего гальванического элемента. На поверхности металла могут быть участки, на которых катодные процессы протекают быстрее (катализируются). Такие участки называют катодными. На других участках будет происходить анодное растворение металла, поэтому они называются анодными участками. Катодные и анодные участки имеют очень малые размеры, однако, они чередуются и образуют коррозионные микроэлементы. Таким образом, при наличии неоднородности поверхности металла коррозионный процесс заключается в работе огромного числа коррозионных микроэлементов. Если металл включения имеет больший потенциал, чем основной металл, то последний становится анодом в образующемся гальваническом микроэлементе и скорость его коррозии возрастает. Так, например, алюминий, содержащий включения железа или меди, коррозирует значительно быстрее, чем алюминий высокой чистоты.
Пример 4.
Атмосферная коррозия алюминия в нейтральной среде протекает по схеме:
(а) 2Al – 6e = 2Al 3+;
(к) 3Н2
О + 3е = 3Н2
+ 3ОН
-
.
Продуктами коррозии являются в данном случае водород и гидроксид алюминия.
Пример 5.
Медь не вытесняет водород из разбавленных кислот вследствие того, что ее потенциал положительнее потенциала водорода. Однако если к медной пластинке, опущенной в кислоту, прикоснуться цинковой пластинкой, то на меди начинается бурное выделение водорода. Это происходит потому, что образуется гальваническая пара, в которой более активный металл (цинк) служит анодом. На аноде происходит окисление цинка по схеме: (а) Zn – 2e = Zn2+
;
На меди, ставшей катодом, происходит восстановление водорода: (к) 2H+
+ 2e = H2
.
Пример 6
: При нарушении целостности медного покрытия на алюминии имеет место коррозия вследствие образования коррозионного гальванического элемента:
(-)2Al/2Al3+
|H2
SO4
|(Cu)3H2
/6H+
(+)
За 45 секунд работы этой гальванопары выделилось 0,008 г водорода. Какая масса алюминия растворилась за это время и какую силу тока даст эта гальванопара?
Силу тока, даваемого коррозионным гальваническим элементом, находим по закону Фарадея (4.8):
 mF
0,008 96500 , mF
0,008 96500 ,
I
17,5a
Эt
45 1
Массу растворившегося за это время алюминия находим также по закону Фарадея (4.8), учитывая, что эквивалентная масса алюминия равна его атомной массе (27) деленной на валентность (3), то есть 9.
 m ЭIt
9 17,5 45 0,072г m ЭIt
9 17,5 45 0,072г
F
96500
Соотношение между потенциалами контактирующих металлов зависит не только от природы металлов, но и от природы растворенных в воде веществ и температуры. Так, в случае контакта железа с цинком последний интенсивно коррозирует при комнатной температуре, но в горячей воде полярность металлов изменяется, и коррозировать начинает железо.
Для защиты от коррозии
и предупреждения ее применяются различные методы. К важнейшим из них относятся следующие методы:
1) Легирование металлов
. При легировании в состав сплава вводят компоненты, вызывающие пассивацию основного металла и повышение его устойчивости к коррозии. В качестве таких легирующих компонентов
применяют хром, никель, вольфрам и другие металлы. Легирование металлов – эффективный, хотя и дорогой способ защиты от коррозии.
2) Защитные покрытия
. Слои различных материалов, создаваемые на поверхности металлических изделий и сооружений для защиты от коррозии называются защитными покрытиями. Материалами для защитных покрытий могут быть как чистые металлы цинк, кадмий, алюминий, никель, медь, хром, серебро, так и их сплавы (бронза, латунь и др.).
Защитные покрытия делятся на катодные и анодные покрытия
. К катодным покрытиям относятся такие металлические покрытия, потенциалы которых имеют более положительное значение
, чем потенциал основного металла. Примерами катодного покрытия на стальных изделиях являются медь, серебро, никель. При повреждении покрытия или при наличии в нем пор возникает коррозионный элемент, в котором основной материал служит анодом и растворяется (коррозирует), а материал – катодом, на котором выделяется водород или поглощается кислород. Таким образом, катодные покрытия могут защищать основной металл от коррозии лишь при отсутствии на нем повреждений или пор.
Анодные покрытия имеют более отрицательный потенциал, чем потенциал основного металла.
Примером анодного покрытия может служить цинковое покрытие на стальных изделиях. При повреждении покрытия анодом будет служить металл покрытия, а основной металл, в качестве катода, разрушению подвергаться не будет. Потенциалы металлов зависят от состава растворов, поэтому, например, покрытие стали оловом (лужение) в растворе серной кислоты является катодным, а в растворе органических кислот – анодным.
Пример 7
. Железное изделие покрыли кадмием. Какое это покрытие – катодное или анодное? Составьте уравнения анодного и катодного процессов коррозии этого изделия во влажном воздухе и в соляной кислоте. Какие продукты коррозии образуются в первом и во втором случае?
Кадмий (потенциал –0,403в) менее активный металл, чем железо, (потенциал –0,440в) и в случае образование коррозионного элемента будет служить катодом, поэтому данное покрытие является катодным. При коррозии происходит анодное растворение железа: (а) Fe – 2e = Fe2+
;
Катодным процессом в случае атмосферной коррозии во влажном воздухе будет восстановление кислорода:
(к) H2
O +O2
+ 4e = 4OH-
;
Продуктами коррозии в данном случае являются гидроксид железа. В кислой среде происходит катодное восстановление ионов водорода:
(к) 2Н+
+2е = Н2
.
Продуктами коррозии в этом случае являются хлорид железа (II) и водород.
3) Электрохимическая защита
. Этот метод защиты от коррозии основан на торможении анодных или катодных реакций коррозионных процессов. К защищаемой конструкции присоединяют металл с более отрицательным электродным потенциалом, чем потенциал металла конструкции. Этот металл называется протектором
, а защита от коррозии – протекторной защитой
. При хорошем контакте защищаемый металл (например, железо) и металл протектора (например, цинк) оказывают друг на друга поляризующее действие в соответствии с их положением в ряду активности металлов. Железо поляризуется катодно, а цинк – анодно. В результате на железе идет процесс окисления того окислителя, который вызывает коррозию (это обычно растворенный в воде кислород), а цинк окисляется. Протекторы широко применяются для защиты морских судов. Ясно, что убытки, связанные с ремонтом громадного судна вследствие коррозии его конструкций во много раз превысили бы стоимость протекторов. Используется также катодная или анодная поляризация за счет приложенного извне тока
. Сущность катодной защиты заключается в том, что защищаемое изделие присоединяется к отрицательному полюсу внешнего источника постоянного тока и становится вследствие этого катодом. Анодом обычно служит стальной вспомогательный электрод, который растворяется. Анодную защиту применяют к металлам, способным легко пассивироваться (образовывать оксидную пленку) при смещении их потенциала в положительную сторону. Анодную защиту применяют, например, для предотвращения коррозии нержавеющих сталей в серной кислоте.
4) Изменение свойств коррозионной среды.
Для снижения агрессивности среды уменьшают концентрацию в ней компонентов, опасных в коррозионном отношении. В нейтральных средах, например, коррозия протекает обычно с поглощением кислорода. Кислород удаляют кипячением или вытеснением его из раствора при помощи инертного газа (барботаж инертным газом) или восстанавливают соответствующими реагентами (сульфиты, гидразин). Агрессивность кислых сред можно снизить подщелачиванием (нейтрализацией). Для защиты от коррозии широко применяют вещества, при добавлении которых в соответствующую среду значительно уменьшается скорость коррозии. Такие вещества называются ингибиторами коррозии
. По составу ингибиторы делятся на органические и неорганические. Так как активность ингибиторов зависит от рН среды, их также делят на кислотные, щелочные и нейтральные. По механизму действия ингибиторы можно разделить на анодные, катодные и экранирующие
. Анодные замедлители, например, нитрит натрия или дихромат калия, тормозят анодные процессы. Катодные замедлители снижают скорость коррозионного процесса за счет снижения интенсивности катодного процесса. К ним относятся такие органические вещества, как диэтиламин, уротропин, формальдегид и пр. Экранирующие ингибиторы (амины с небольшой молекулярной массой с добавлением группы -NO3
или -СО3
) адсорбируются на поверхности металла, предохраняя его от контакта с агрессивными средами, вызывающими коррозию металла.
4.4. Вопросы и задания:
1. Как измеряют электродные потенциалы? Что такое водородный электрод?
2. Чем отличается электролиз расплавов от электролиза растворов?
3. Сформулируйте объединенный закон электролиза Фарадея. Что такое выход по току?
4. Что такое электрохимическая коррозия?
5. Определите значение электродного потенциала меди, погруженной в раствор азотнокислой меди Cu(NO3
)2
концентрацией 0,0005М.
6. Составьте схему работы гальванического элемента, образованного железом и свинцом, погруженными в 0,005М растворы их солей. Рассчитайте ЭДС этого элемента и изменение величины энергии Гиббса.
7. Составьте уравнения катодного и анодного процессов, протекающих при электролизе водного раствора бромида меди CuBr2
.
8. Через раствор нитрата никеля в течение 2,45 часа пропускали ток силой 3,5А. Определите, на сколько граммов за это время уменьшилась масса никелевого анода.
9. Как происходит атмосферная коррозия луженого железа и луженой меди при нарушении покрытия? Составьте электронные уравнения катодного и анодного процессов.
10. Железо покрыли никелем. Какое это покрытие – анодное или катодное? Какой металл будет коррозировать при нарушении покрытия в кислой среде? Составьте уравнения катодного и анодного процессов.
Лекция 5. Катализаторы и каталитические системы
То, что при кипячении раствора крахмала происходит его медленное превращение в сахар, было известно давно. Но в начале прошлого века русский химик К.С.Кирхгоф обнаружил, что реакция заканчивается практически мгновенно, если в раствор добавить серную кислоту. Английский химик Х.Дэви открыл свойство платины многократно ускорять процесс окисления метана. Самым удивительным было то, что ни серная кислота, ни платина не изменялись в ходе реакции. Дэви и Кирхгоф открыли явление катализа,
хотя сущность этого удивительного явления была понята много позже. Наиболее точное определение катализа принадлежит академику Г.К. Борескову: «
Катализом называется ускорение химических реакций в присутствии определенных веществ, многократно химически взаимодействующих с реагентами (исходными веществами), но не входящих в состав продуктов
реакции».
Вступающий в реакцию реагент называют субстратом. Катализатором
называют вещество, ускоряющее реакцию. Ингибитором
называют вещество, замедляющее реакцию. Селективностью
катализатора называют его свойство ускорять преимущественно одну из нескольких реакций. Промоторами
называют вещества, увеличивающие эффективность катализатора. Каталитическим ядом
называется примесь, снижающая эффективность катализатора.
Различают гомогенный и гетерогенный катализ
. В случае гомогенного катализа катализатор и реагирующие вещества образуют одну фазу (газ или раствор). В случае гетерогенного катализа катализатор находится в системе в виде самостоятельной фазы. Примером гомогенного катализа является каталитическое разложение пероксида водорода на воду и кислород в присутствии дихроматов, молибдатов или вольфраматов. Константа скорости реакции в этом случае определяется как
К
кат
 С
к
, (5.1) С
к
, (5.1)
где Ak
– производительность катализатора, то есть выход продукта с 1 кг; Ск
– его концентрация.
Гетерогенные каталитические системы
применяют, например, при окислении диоксида серы до триоксида при производстве серной кислоты, при синтезе аммиака или его окислении при производстве азотной кислоты. При гетерогенном катализе определяющим фактором является время контакта:
М
прод
 А
л
(5.2) t m
кат А
л
(5.2) t m
кат
где Ak
– производительность катализатора; Mпрод
– масса продукта; t –
время контакта; mкат
– масса катализатора.
Для реакций, которые катализируются по механизму гетерогенного катализа, в 1913 году было выведено кинетическое уравнение Михаэлиса-Ментен для стационарной скорости реакции:
 V
max [S
] (5.3) V
max [S
] (5.3)
V
ст
K
m
[S
]
где KM
– константа Михаэлиса, vmax
– максимальная скорость, S – площадь поверхности катализатора.
5.1. Механизм действия катализаторов
Механизм каталитического процесса настолько сложен, что до сих пор не удалось создать общую теорию, объясняющую ускоряющее действие катализаторов. Большинство гомогенных
каталитических процессов можно удовлетворительно объяснить при помощи теории промежуточных соединений
. Первое теоретическое объяснение было предложено Оствальдом в 1894-1911 гг. Основное положение этой теории состоит в том, что в течение реакции образуются неустойчивые промежуточные соединения катализатора с реагирующими веществами, которые затем распадаются с образованием продуктов реакции, а катализатор регенерируется. При реакции между веществами А и В с участием катализатора К на первом этапе образуется промежуточное соединение А с катализатором К, которое затем реагирует с В с выделением катализатора в свободном виде:
1) А + К = АК; 2) АК + В = АВ + К
Например, реакция разложения уксусного альдегида:
CH3
CHO = CH4
+ CO
катализируется парами йода и протекает в две стадии:
1. СН3
СНО + J2
= СН3
J +HJ + CO 2. CH3
J + HJ = CH4
+ J2
Строго говоря, добавление катализатора не ускоряет протекание реакции, а открывает иной по механизму, более быстрый путь протекания реакции. Если сравнить протекание химической реакции переходу через горный хребет, то можно сказать, что лишь малой доле самых активных молекул удаѐтся при каждом штурме преодолевать этот перевал. А катализатор, как опытный проводник, ведет группу другим путем, через несколько более пологих перевалов. Этот путь длиннее, но скорость такого перехода, не требующего штурмовых усилий, оказывается гораздо выше. (См. Рис. 5.1).
Рис. 5.1. Ход реакции без катализатора и с катализатором

Как видно из Рис. 5.1 основным эффектом влияния катализатора на протекание реакции является снижение энергии активации[10]
катализируемого процесса.
Существуют также автокаталитические процессы
, скорость которых по мере расхода реагентов не уменьшается, а возрастает.
Например, при реакции этилацетата с водой
СН3
ООС2
Н5
+ Н2
О = СН3
СООН + С2
Н5
ОН
Образующаяся при реакции уксусная кислота является катализатором прямой реакции. Тут нет противоречия с законом действующих масс, поскольку механизм этого явления таков, что промежуточные продукты или продукты реакции оказывают каталитическое влияние на процесс. Эти процессы носят название процессов с положительной обратной связью
. Известны процессы и с отрицательной обратной связью. Если продукт реакции является ее ингибитором (
замедлителем), то скорость реакции будет уменьшаться быстрее, чем уменьшаются концентрации реагентов. Механизмы протекания реакций могут быть разными, однако одно остается неизменным – это обязательное образование и распад неустойчивого соединения, содержащего катализатор.
Одним из современных объяснений механизма гетерогенного катализа является мультиплетная теория
. Она предложена в 1929 году Баландиным. В основе мультиплетной теории лежит принцип структурного и энергетического соответствия
между поверхностью катализатора и молекулами реагирующих веществ. Структурное соответствие заключается в том, что расстояние между атомами кристаллической решетки катализатора и валентные углы между ними должны соответствовать тем же параметрам участвующих в реакции молекул. В этом, и только в этом случае молекулы реагирующих веществ поглощаются катализатором (совершают активированную адсорбцию на катализаторе) с образованием множественных связей между атомами катализатора и молекулами реагентов. Например, расстояние между атомами Ni-Ni на поверхности никеля Ренея
наилучшим образом подходят к ацетиленовой связи, тогда как к этиленовой связи в равной мере подходят межатомные расстояния многих металлов от платины до железа. Вот почему никель Ренея обладает уникальной селективностью по отношению к реакциям гидрирования ацетиленовых углеводородов, а металлы платиновой группы используются при получении соединений этилена.
Атомы поверхности катализатора, принимающие участие в активированной адсорбции, входят в состав мультиплета
. Поверхностное соединение, образующееся в результате активированной адсорбции молекул реагирующих веществ на мультиплете, называется мультиплетным комплексом
. Комплекс представляет собой некий континуум (сплошность), физическое состояние вещества в момент, когда исходные вещества прекратили свое существование, а молекулы продуктов реакции еще не образовались. Мультиплетный комплекс распадается с образованием с образованием продуктов реакции, если этот процесс согласуется с законами термодинамики.
Принцип энергетического соответствия
определяет требования к тому, чтобы адсорбционный потенциал катализатора по возможности ближе подходил к полной энергии реагирующих связей – средней величине из разрываемых и вновь образуемых связей. Энергетическое соответствие катализатора определяется экспериментально, поэтому мультиплетная теория не в состоянии однозначно предсказывать катализатор для той или иной реакции. В гетерогенном катализе участвует лишь поверхность твердого катализатора, поэтому для более интенсивного его использования и в целях экономии стремятся увеличить поверхность катализатора путем его измельчения. Применяется также нанесение катализатора тонким слоем на тело, обладающее сильно развитой поверхностью. Такие материалы (активированный уголь, керамзит, пемза, асбест, силикагель и пр.) называют носителями, или трегерами
.
5.2. Особенности каталитических систем
Селективность катализаторов
позволяет ускорять одну или несколько нужных реакций и «заглушать» протекание нежелательных процессов. Скорость гомогенно-каталитических реакций зависит от концентрации катализатора и температуры. Следовательно, ускорение процесса достигается при увеличении концентрации катализатора и повышении температуры системы, а замедление – обратными процессами. В гетерогенно-каталитическом процессе факторами, позволяющими управлять процессами, являются изменение температуры, интенсивности перемешивания системы и степень измельчения катализатора. Мерой активности катализатора является изменение скорости химической реакции в результате введения в систему катализатора. Количественно активность катализатора оценивается производительностью катализатора Ак
. Под производительностью катализатора подразумевают количество вещества M, получающегося в единицу времени с единицы площади поверхности (Sк
), массы (Mк
) или объема катализатора (Vк
).
Производительность катализатора равна
А
к
 (5.4) (5.4)
Основные особенности каталитических систем можно охарактеризовать следующим образом:
1. Катализатор принимает непосредственное участие в химическом процессе, изменяя его механизм и скорость.
2. Катализатор снижает энергию активации реакции, что и является причиной ускорения реакции.
3. Катализатор в конце процесса, претерпевая термодинамический цикл, возвращается в исходное состояние. Это значит, что катализатор не влияет на функции состояния реагирующей системы, в частности, на тепловой эффект реакции.
4. Катализатор не влияет на положение термодинамического (и химического) состояния равновесия системы, а всего лишь ускоряет установление этого равновесия.
5. Для каждой реакции существуют определенные вещества или смесь веществ, обладающих селективным каталитическим действием. Поэтому, называя катализатор, следует обязательно указывать реакцию или тип реакции, на которые распространяется действие катализатора.
5.3. Перспективы развития каталитических систем
Роль каталитических систем, как в промышленности, так и в окружающем нас мире вообще трудно переоценить. Ускорение множества промышленных процессов невозможно без применения катализаторов. В результате биохимической эволюции миллионы лет шло совершенствование природных катализаторов. Сама жизнь зародилась на Земле благодаря каталитическим процессам. Природные катализаторы – тысячи энзимов
, или ферментов
– ускоряют превращения веществ в живых организмах, в том числе и в организме человека. Среди других видов каталитических систем
ферментативный катализ является самым высокоорганизованным, так как ферменты отличаются высокой избирательностью, селективностью, специфичностью и каталитической активностью. Молекула белковой молекулы-фермента образует активный каталитический центр, содержащий функциональные группы, способные ориентировать молекулы реагирующих веществ в определенном положении к активному центру. В состав активного центра многих ферментов входят ионы металлов, причем при удалении такого иона молекула теряет каталитические свойства. Активные центры имеют строго определенную структуру, что позволяет ферменту присоединять только молекулы определенного строения. Так, например, фермент уреаза гидролизует карбамид (мочевину) в 1014
раз быстрее, чем ион водорода и не оказывает влияния на реакции гидролиза других соединений. В настоящее время известны тысячи ферментов, одни из которых катализируют только окислительно-восстановительные процессы, другие – реакции гидролиза и т.д.
Катализ является одним из самых современных и перспективных направлений химии. Выдающийся химик Н.Н. Семенов представлял химические процессы, протекающие в органах и тканях растений, животных и человека, в виде химического производства живой природы. По его мнению, в ходе эволюции организмов природа создала «молекулярные машины» исключительной точности, быстроты действия и необычайного совершенства. В качестве примера можно привести матричный синтез белковых молекул (см. Лекцию 18). Клетки живых организмов содержат субмикроскопические сборные «заводики» - рибосомы, содержащие «сборные машины» - транспортные и информационные рибонуклеиновые кислоты. Каждый вид коротких молекул транспортных рибонуклеиновых кислот захватывает один определенный тип аминокислот, несет их в рибосому и ставит на свое место в соответствии с информацией, содержащейся в информационной РНК. Тут же к аминокислотам подходят катализаторыферменты и производят «сшивание» аминокислот в макромолекулу белка, соблюдая их строгую очередность. Это настоящий завод, строящий молекулу белка по плану, выработанному в ходе эволюции. Многие ферментативные реакции протекают через стадию образования между субстратом (реагентом, вступающим в реакцию) S и ферментом (катализатором) E промежуточного фермент-субстратного комплекса SE, который затем распадается с регенерацией фермента E и образованием продукта P:
1) S + E = SE; 2) SE = E + P
Используя принципы химии организмов, можно построить совершенно новую, эволюционную химию
, основанную на необычном управлении химическими процессами при помощи биокатализаторовферментов
. Установлены важные положения эволюционной химии:
аналогия между химическим катализом и биокатализом, между ферментами и катализаторами; наличие в ферментах активных центров и носителей; заключение о важной роли металлоорганических соединений в биокатализе; вывод о распространении на биокатализ законов химической кинетики; сведение (в отдельных случаях) биокатализа к катализу неорганическими агентами. В настоящее время эволюционная химия уровня живой лаборатории еще не достигла, но наметились три подхода к освоению опыта живой природы:
 Катализ при помощи металлоорганических соединений,
объединяющий методы классического гетерогенного катализа с приемами, которыми пользуются живые организмы в ферментативных реакциях. Первые шаги в этом направлении сделаны в 1954 году немецким химиком, лауреатом Нобелевской премии К. Циглером
(1898 -1973), который открыл реакцию полимеризации олефинов (например, этилена) при помощи катализатора на основе металлоорганических соединений титана и алюминия (известной сейчас под названием реакции Циглера-Натта). В настоящее время существует более 40 промышленных процессов с участием металлоорганических катализаторов. Катализ при помощи металлоорганических соединений,
объединяющий методы классического гетерогенного катализа с приемами, которыми пользуются живые организмы в ферментативных реакциях. Первые шаги в этом направлении сделаны в 1954 году немецким химиком, лауреатом Нобелевской премии К. Циглером
(1898 -1973), который открыл реакцию полимеризации олефинов (например, этилена) при помощи катализатора на основе металлоорганических соединений титана и алюминия (известной сейчас под названием реакции Циглера-Натта). В настоящее время существует более 40 промышленных процессов с участием металлоорганических катализаторов.
 Второй путь заключается в моделировании биокатализаторов. Второй путь заключается в моделировании биокатализаторов.
Искусственно отбирая структуры, удалось построить модели многих ферментов с высокой активностью и селективностью, но с более простым строением, чем у природных биокатализаторов. Но ни одна полученная модель до сих пор не смогла заменить природную систему. Дело в том, что такой искусственный катализатор работает всего несколько минут, во время работы он разрушается (как любой «классический» катализатор). В живых организмах ферменты способны самовосстанавливаться и самосовершенствоваться.
Третий путь называется химией иммобилизированных систем.
Благодаря успехам микробиологии стало возможно получать дешевые ферменты, которые в 100 - 1000 раз дешевле натуральных продуктов. Но главное, найдены пути стабилизации ферментов и создания иммобилизированных систем. Сущность иммобилизации
состоит в закреплении выделенных из живого организма ферментов на твердой поверхности путем адсорбции. При этом ферменты превращаются в гетерогенный катализатор, который может работать непрерывно и стабильно. Больших успехов в этой области достиг И.В. Березин (1923 - 1987) и его сотрудники из Московского университета. Некоторые процессы уже реализованы в промышленных масштабах.
Четвертый путь – это изучение и освоение всего каталитического опыта живой природы
, в том числе и формирования самого фермента, клетки и даже самого организма.
5.4. Вопросы и задания
1. Объясните механизм действия катализатора на основе теории промежуточных соединений.
2. Что такое автокатализ?
3. Какой принцип лежит в основе мультиплетной теории гетерогенного катализа?
4. Что такое катализатор Ренея?
5. Какие каталитические системы называют системами с положительной обратной связью? С отрицательной обратной связью?
6. Как изменится константа скорости катализируемой реакции при гомогенном катализе, если заменить катализатор другим, активность которого в 10 раз выше, и уменьшить его концентрацию в 5 раз?
7. Найдите объѐм катализатора для синтеза аммиака NH3, если производительность установки составляет 5000 м3
/час.
Производительность катализатора равна 2000 кг/(м3
∙час).
8. При окислении аммиака на платиновом катализаторе было получено в течение суток 1440 кг азотной кислоты. Для окисления использовано 0,064 кг катализатора. Рассчитайте активность катализатора.
9. Какую реакцию ускоряет катализатор Циглера-Натта?
10. Что такое эволюционная химия?
Лекция 6. Полимеры и олигомеры
Полимеры и олигомеры относятся к органическим соединениям.
6.1. Органические соединения
Органические соединения – это
соединения углерода (за исключением самых простых, как, например, как оксид и диоксид углерода, карбиды, и пр., которые считаются неорганическими соединениями), так как в природе они встречаются почти исключительно в организмах животных и растений, принимают участие в процессах жизнедеятельности или распада организмов. Деление веществ на органические и неорганические условно, так как множество соединений углерода можно обнаружить как в живой, так и неживой природе. В 1824 году немецкий химик Ф.Вѐлер получил из неорганического вещества дициана C2
N2
органическую щавелевую кислоту HOOC-COOH, которую до тех пор добывали только из растений. В 1928 году он же осуществил первый синтез органического вещества животного происхождения: нагреванием цианата аммония NH4
CNO получил мочевину, или карбамид (NH2
)2
CO. В настоящее время путем органического синтеза
получают множество органических соединений. Современная теория органических соединений основывается на следующих положениях:
Все особенности органических соединений, прежде всего, определяются свойствами элемента углерод. Электронная формула углерода (1s2
)2s2
2p2
;
в возбужденном состоянии углерод имеет электронную конфигурацию 2s1
2p3
и может образовывать четыре ковалентные связи. Из одной s- и трех р-орбиталей в результате гибридизации образуются четыре симметричные sp3
-орбитали, которые в результате перекрывания с электронными облаками других атомов (например, с 1s-шаровыми облаками атомов водорода) образуют так называемые ковалентные сигма-связи.
Исключительным свойством углерода является способность его атомов соединяться друг с другом прочными ковалентными связями.
При этом образуются углеродные цепи практически неограниченной длины. Свободные валентности используются для присоединения других атомов или групп. Таким образом, можно выделить ряды однотипных органических соединений (гомологические ряды), в которых каждый последующий член отличается от предыдущего на один атом углерода. Так, например, если насыщать свободные валентности углеродных цепей атомами водорода, получится гомологический ряд насыщенных, или
предельных углеводородов (метан, этан, пропан, бутан и т.д.)
Связи между атомами углерода в цепи могут быть кратными – двойными или тройными
. Такие связи образуются двумя или тремя общими парами электронов двух соседних атомов углерода. Такие углеводороды называются непредельными. Кратные связи при реакциях легко превращаются в простые, поэтому непредельные углеводороды очень реакционноспособны.
Цепи из углеродных атомов могут быть разветвленными
. Так, состав С5
Н12
имеют три предельных углеводорода – пентан и два изопентана (третичный и четвертичный). Такие соединения называются изомерами. Известно несколько видов пространственной изомерии (или стереоизомерии): зеркальная изомерия асимметрического атома углерода; цис-транс-изомерия, изомерия положения кратной связи и пр. Многообразие органических молекул за счет пространственной симметрии определяется также свойством углеродных цепей замыкаться в кольцо. Самым известным соединением такого рода является бензол; бензольное кольцо - энергетически очень устойчивая структура. Существуют также молекулы, замкнутые в цикл из пяти углеродных и других атомов.
Непосредственно связанные друг с другом атомы органического соединения взаимно влияют друг на друга
. Атомы или группы атомов, замещающие водород в углеводородной основе, образуют функциональные группы. Органические вещества, имеющие одинаковые функциональные группы, принадлежат к одному и тому же классу. Ниже приведены формулы и названия некоторых функциональных групп и названия соответствующих классов органических соединений.
Гидроксильная
группа - OH
входит в состав спиртов (метанола, этанола, пропанола и т.д.);
Карбонильная
группа - COH
входит в состав альдегидов и кетонов (ацетона, формалина);
Карбоксильна
я
группа - COOH
образует класс карбоновых кислот (муравьиной, уксусной, пропионовой, масляной и т.д.);
Аминогруппа
- NH2
входит в состав аминов (например, метиламина, гексаметилендиамина) и аминокислот;
Нитрильная группа
- СN
входит в состав нитрилов.
Разнообразные сочетания углеродных цепей различной пространственной конфигурации с разнообразными функциональными группами обеспечивает многообразие органических соединений, которое лежит в основе многообразия форм живой материи на ее молекулярном уровне.
У органических молекул есть еще одно очень важное свойство: в соответствующих условиях простые молекулы – мономеры
,
способны соединяться вместе, образуя одну громадную макромолекул
у
, состоящую из большого числа звеньев. Такие молекулы называют полимерами
от греческих слов polus
– много и meris
– часть. Молекулы полимеров имеют молекулярную массу порядка сотен, тысяч и даже миллионов единиц относительной атомной массы. Полимеры выделяют в особую группу органических соединений под названием «высокомолекулярные соединения» (ВМС).
Поскольку кремний является химическим аналогом углерода, на основе кремниевых соединений также можно получить полимерные молекулы, которые называют неорганическими полимерами
. Высокомолекулярные соединения делятся на природные ВМС и синтетические ВМС.
6.2. Природные полимеры
К природным ВМС
относятся крахмал и целлюлоза
, натуральный каучук
, белки
, нуклеиновые кислоты
ДНК и РНК и многие другие природные соединения.
Крахмал и целлюлоза
являются высокомолекулярными несахароподобными углеводами с общей формулой (С6
Н10
О5
)n. Мономерными звеньями этих ВМС являются остатки глюкозы. Крахмал состоит из остатков α-глюкозы, а целлюлоза – из β-глюкозы, которые являются пространственными изомерами и отличаются лишь положением одной гидроксильной группы. Крахмал накапливается в некоторых растениях в виде резервного полисахарида. Из целлюлозы построены ткани растений. Вата и фильтровальная бумага более чем на 95% состоят из целлюлозы. При полном гидролизе крахмала, то есть реакции его с водой при нагревании и в присутствии катализатора, серной кислоты, образуется α-глюкоза. При полном гидролизе целлюлозы получают β-глюкозу.
Каучук натуральный,
полимер растительного происхождения, вулканизацией которого получают резину, относится к группе эластомеров, то есть высокомолекулярных соединений, обладающих способностью к большим обратимым деформациям при обычных температурах. Добывают каучук, главным образом, из латекса бразильской гевеи, которая культивируется в тропических странах. В 1738 французский исследователь Ш. Кондамин представил в Академию наук Париже образцы каучука, изделия из него и описание способов добычи в странах Южной Америки. Промышленное применение натурального каучука стало возможным после открытия процесса вулканизации. Основная составная часть натурального каучука (91— 96%) – это полиизопрен (C5
H8
) n
, то есть полимер изопрена СН2
= С (СН3
) – СН = СН2
, где число мономерных звеньев состава (- СН2
– С (СН3
) = СН – СН2
-) составляет от 1400000 до 2600000. Кроме того, натуральный каучук содержит также 2,2—3,8% белков и аминокислот, 1,5—4,0% веществ, извлекаемых ацетоном (так называемый ацетоновый экстракт — олеиновая, стеариновая, линолевая кислоты, каротин и др.), соединения металлов переменной валентности — меди (до 0,0008%), марганца (до 0,001%), железа (до 0,01%), песок и некоторые др. примеси. Натуральный каучук является стереорегулярным полимером
; 98—100% звеньев изопрена в его макромолекуле соединяются в положении 1,4 цис:

Ценным свойством натурального каучука является его высокая когезионная прочность, именно благодаря этому свойству каучук незаменим в производстве деталей шин. Технологическим недостатком натурального каучука, - вследствие его высокой молекулярной массы, — является необходимость пластикации перед введением составных частей резиновой смеси. Наиболее распространѐнным вулканизующим агентом для натурального каучука является сера; в качестве ускорителей вулканизации применяют 2-меркаптобензтиазол (каптакс) и другие катализаторы. Резины из натурального каучука обладают хорошей эластичностью, высокой износостойкостью и морозоустойчивостью, однако подвержены воздействию растворителей, масел, а также отличаются меньшей, чем у некоторых синтетических каучуков, термостойкостью и устойчивостью к воздействию атмосферных осадков. Основная область применения натурального каучука – это производство шин. Его используют также в производстве резинотехнических изделий (транспортѐрные ленты, приводные ремни, амортизаторы, уплотнители), электроизоляционных материалов, резиновых изделий народного потребления, при изготовлении резиновых клеев. Некоторое количество натурального каучука в виде латекса. Благодаря созданию стереорегулярных синтетических каучуков, а также широкого ассортимента синтетических каучуков специального назначения, потребление натурального каучука в промышленности сокращается.
Человеческий организм состоит из десятков тысяч различных белков, каждый из которых имеет собственную структуру и функции. Белковые молекулы – самые сложные молекулярные структуры, известные на сегодня. У каждого типа белка имеется уникальная трехмерная структура. Но при всем своем разнообразии все белки
являются полимерными молекулами, состоящими из универсального набора из 20 аминокислот
– мономеров, которые называют «каноническими».
В органической химии известно несколько сотен аминокислот, однако в состав всех
белков, из которых состоят живые существа, входят только эти 20 аминокислот
. Это глицин (Gly); аланин (Ala); валин* (Val); лейцин* (Leu); изолейцин* (Ile); метионин (Met); фенилаланин (Phe); триптофан (Trp); пролин (Pro); Серин (Ser); треонин* (Thr); цистеин (Cys); тирозин (Tyr); аспарагин (Asn); глутамин (Gln); аспарагиновая кислота (Asp); глютаминовая кислота (Glu); лизин* (Lys); аргинин (Arg); гистидин (His). В скобках приведены принятые сокращения для названия аминокислот, значком * обозначены незаменимые аминокислоты, которые не могут быть синтезированы организмом из более простых веществ.
Большинство аминокислот содержат асимметричный атом углерода (альфа-углерод), связанный с четырьмя разными партнерами ковалентной связью. Каждая аминокислота содержит карбоксильную группу и аминогруппу, соединенную с атомом альфа - углерода. Общую формулу аминокислот можно представить в следующем виде: NH2
– CHR –COOH, где R – это углеводородный радикал простого (например, Н у глицина, СН3
у аланина) или сложного состава.
Поскольку атом альфа-углерода несимметричен, каждая аминокислота может существовать в виде двух
оптических изомеров: L (α)
– форме (от латинского слова laesus
- левая) и D (β)
- форме (от латинского слова dexter
- правая). При синтезе аминокислот в лабораторных условиях получают рацемическую смесь обеих форм, но молекулы природных белков
, за редким исключением, состоят из аминокислот в левовращающей (α) - форме.
При синтезе белка молекулы аминокислот располагаются таким образом, что карбоксильная группа одной кислоты сближается с аминогруппой другой кислоты. В результате реакции дегидратации происходит синтез макромолекулы, содержащей пептидные связи.
NH2
-C(R1
H)-COOH + NH2
–C(R2
H)-COOH = NH2
-C(R1
H)-CO-NH-C(R2
H)–COOH + H2
O
На концах макромолекулы располагаются с одной стороны аминогруппа, а с другой - карбоксильная группа. Все остальные связываются пептидными связями. Повторяющаяся последовательность атомов азота и углерода в такой цепи называется полипептидным скелетом.
К скелету прикреплены различные группы атомов, образующие боковые цепи. Полипептидные цепи могут быть короткими, состоящими всего из нескольких мономеров, а могут состоять из тысяч и тысяч звеньев. Каждая разновидность белка имеет уникальную последовательность линейных мономеров.
Так, например, полипептидная цепь лизосомы, относительно простого белка-энзима, представляет собой цепочку из 129 аминокислот. Даже незначительное нарушение порядка расположения аминокислот в полипептидной цепи влияет на свойства и функции белка. Например, болезнь крови под названием анемия возникает из-за того, что одна аминокислота в полипептидной цепочке заменяется другой. Определенная последовательность аминокислот, определяющая первичную структуру белка, задается наследственной генетической программой. На рубеже 40-х и 50-х годов XX века Фредерик Сангер, специалист по молекулярной биологии из Кембриджского университета в Англии, впервые определил последовательность аминокислот в молекуле инсулина, доказав тем самым, что свойства белка определяются, в первую очередь, последовательностью аминокислот в первичной структуре белка.
Молекула любого белка состоит из одной или нескольких полипептидных цепей (первичной структуры
), которая скручивается и сматывается в клубок, образуя макромолекулу строго определенной пространственной конфигурации с вторичными, третичными и четвертичными структурами. Вторичная
структура белка (спиралевидная или имеющая форму «плиссированного листа») возникает за счет водородных связей между звеньями пептидов. Третичная
структура менее правильна по форме, она образуется за счет химических связей в боковых цепях с гидрофобными функциональными группами. Эти связи образуют водородные или сульфидные «мостики». Четвертичная
структура характерна для белков, в состав которых входит более одной пептидной цепи.
В естественных условиях конфигурация молекулы белка устойчива и определяет все его функции. Если же условия окружающей среды (температура, рН, концентрация солей) изменяются, происходит денатурация
белка, и он не может больше выполнять свои функции. При изменении условий окружающей среды денатурированный белок может вернуться в исходное состояние.
Функции же белков разнообразны. Они зависят от способности белков узнавать и присоединять различные молекулы. Это свойство белков называется биологическим узнаванием
.
Например, белки антител,
например, интерферон,
узнают и связывают определенные чужеродные тела, которые наносят вред организму. Гемоглобин
присоединяет кислород и разносит его от легких по всему телу, а инсулин
регулирует концентрацию сахара в крови.
Существует два типа нуклеиновых кислот –
дезоксирибонуклеиновая кислота (ДНК)
и рибонуклеиновая кислота (РНК). Обе они являются теми молекулами, посредством которых живые организмы способны воспроизводиться – создавать новые организмы, повторяющие из поколения в поколение сложную структуру и свойства каждого живого существа. Нуклеиновые кислоты состоят из мономеров, которые называются нуклеотидами. Катализаторы – энзимы сшивают их в цепи в реакциях дегидратационного синтеза. Каждый нуклеотид, в свою очередь, состоит из трех частей: азотистого основания, соединенного с пентозой (полисахаридом с цепью из пяти атомов углерода), которая в свою очередь связана с фосфатной группой. Азотистые основания бывают двух типов: пиримидиновые (шестичленное кольцо из атомов углерода и азота) и пуриновые (пятичленное кольцо, соединенное с пиримидиновым шестичленным кольцом). Азот пуринов и пиримидинов способен отбирать ион водорода у молекулы воды, поэтому эти молекулы являются азотистыми основаниями. Пентозы, входящие в состав нуклеотидов, являются оптическими изомерами в d(β) –форме. Асимметрия молекул живых систем называется молекулярной хиральностью
. Химический состав нуклеотидов в нуклеиновых кислотах приведен в Таблице 6.1.
Таблица 6.1. Химический состав нуклеиновых кислот
Состав ДНК РНК
Пурины Аденин (А) Аденин (А)
Гуанин (Г) Гуанин (Г)
Пиримидины Цитозин (Ц) Цитозин (Ц) Тимин (Т) Урацил (У)
Пентозы (сахара) Дезоксирибоза Рибоза
Кислотный остаток Фосфатная группа Фосфатная группа
Поскольку нуклеотиды отличаются друг от друга только входящими в их состав азотистыми основаниями, названия азотистых оснований служат названиями нуклеотидов.
ДНК является уникальной молекулой, которая способна воспроизводить сама себя, и этот механизм наследственности на молекулярном уровне лежит в основе жизни. Каждый раз, когда делится клетка, ее ДНК копируется и передается от одного поколения клеток к другому. Нуклеиновая кислота второго типа, РНК, участвует в синтезе белков в соответствии с информацией, содержащейся в ДНК. Молекула РНК представляет собой полимерную цепочку, состоящую из нуклеотидов аденина (А), гуанина (Г), цитозина (Ц) и урацила (У).
Молекула ДНК представляет собой двойную спираль, обе нити которой связаны водородными связями азотистых оснований. Каждая отдельная связь достаточно слаба, но все вместе они образуют связь, подобную той, которую обеспечивает застежка-молния. Нуклеотиды каждой нити связаны меж собой прочными ковалентными связями. Схема строения молекулы ДНК приведена на Рис. 6.1.
Рис. 6.1. Строение молекулы ДНК
 Молекулы ДНК сложных живых систем очень длинны. Так, в каждой клетке организма человека содержится почти 2-метровая молекула ДНК, упакованная в 46 хромосом. Молекула ДНК состоит из огромного количества генов
– отдельных участков громадной полимерной молекулы, каждый из которых хранит информацию о первичной структуре Молекулы ДНК сложных живых систем очень длинны. Так, в каждой клетке организма человека содержится почти 2-метровая молекула ДНК, упакованная в 46 хромосом. Молекула ДНК состоит из огромного количества генов
– отдельных участков громадной полимерной молекулы, каждый из которых хранит информацию о первичной структуре
одной белковой молекулы. Организм человека состоит из 30000 разновидностей белковых молекул.
Не всякие сочетания азотистых оснований возможны при образовании ДНК. Существуют пары, всегда дополняющие друг друга по принципу комплиментарности
. Так, аденин
(А) всегда соединяется с тимином
(Т), а гуанин
(Г) с цитозином
(Ц). Если бы мы смогли узнать точно всю последовательность мономеров, входящих в состав одной половины спирали молекулы ДНК, мы бы автоматически узнали и состав другой половины. Например, цепи АГГТЦЦГ
всегда соответствовала бы цепь ТЦЦАГГЦ
, так как обе части спирали ДНК дополняют одна другую. Именно это свойство ДНК делает возможным точное воспроизведение наследственной генной структуры.
6.3. Синтетические полимеры
Синтетические полимеры можно разделить на три основные группы:
1. Пластические массы (пластмассы)
– высокомолекулярные вещества, подвергающиеся пластической обработке. Пластмассы, которые подвергаются пластической обработке только при нагревании, называются термопластичными пластмассами, или термопластами
. К ним относятся поливинилхлорид, который получается при полимеризации винилхлорида СН2
=СHCl
; полиэтилен, полистирол, образующийся при полимеризации стирола СН2
= СН (С6
Н5
)
, и др.
Пластичные на стадии обработки пластмассы, которые в результате последующего воздействия на них (термического или химического) становятся твердыми и неплавкими, называются термореактивными пластмассами
. Это так называемые фенопласты и аминопласты.
2. Эластомеры (синтетические каучуки и резины) – синтетические ВМС, обладающие резиноподобными свойствами. Сырой каучук липок, непрочен и при понижении температуры становится хрупким. Для придания каучукам прочности и эластичности их подвергают вулканизации (вводят серу и нагревают). Вулканизированный каучук называется резиной. В результате многолетней работы академика
С.В.Лебедева был разработан способ получения искусственного каучука, а в 1932 году было налажено его промышленное производство. В разработке метода получения синтетического каучука Лебедев пошѐл по пути подражания природе. Поскольку натуральный каучук является полимером диенового углеводорода изопрена, то Лебедев воспользовался также диеновым углеводородом, только более простым и доступным – бутадиеном (дивинилом). Сырьѐм для получения бутадиена служит этиловый спирт:
С2
Н5
ОН → СН2
=СН-СН=СН2
→(-СН2
-СН=СН-СН2
-)n
.
Химическая промышленность производит много различных видов синтетических каучуков: полибутадиеновый (СКБ), который получают полимеризацией бутадиена СН2
=СН–СН=СН2
по методу Лебедева; бутадиен-стирольный (СКС) и бутадиен-нитрильный (СКН) каучуки, где звенья бутадиена чередуются соответственно со звеньями стирола СН2
= СН (С6
Н5
)
и акрилонитрила СН2
=СН–С=N
(нитрила акриловой кислоты). Получают и синтетический полиизопреновый каучук (СКИ), близкий по свойствам к натуральному природному каучуку.
3. Химические волокна
– синтетические ВМС, сформованные в виде нитей (волокон) и используемые для изготовления текстильных изделий. Различаю две формы химических волокон – непрерывное волокно (искусственный шелк или корд) и штапельное волокно (коротко разрезанные волокна-штапельки для изготовления тканей типа ХБ и шерстяных). Различают полиакрилонитрильные волокна
(орлон, нитрон, дралон, вольприла); полиамидные волокна
(дедерон, найлон, перлон, капрон); полиэфирные волокна
(терилен, лавсан, элана).
Синтетические высокомолекулярные соединения получают двумя способами. Это:
 1. Реакция полимеризации
– образование высокомолекулярных соединений из низкомолекулярных без выделения побочных низкомолекулярных продуктов. Мономерами для реакции полимеризации служат соединения с кратными связями (двойными или тройными). В процессе полимеризации происходит разрыв кратных связей и возникновение химических связей между мономерами с образованием макромолекулы. Так, например, полимеризуется этилен с образованием полиэтилена: nСН2
= CH2
(- CH2
– CH2
- )n 1. Реакция полимеризации
– образование высокомолекулярных соединений из низкомолекулярных без выделения побочных низкомолекулярных продуктов. Мономерами для реакции полимеризации служат соединения с кратными связями (двойными или тройными). В процессе полимеризации происходит разрыв кратных связей и возникновение химических связей между мономерами с образованием макромолекулы. Так, например, полимеризуется этилен с образованием полиэтилена: nСН2
= CH2
(- CH2
– CH2
- )n
Если макромолекула состоит из одинаковых молекул мономера, имеет место гомополимеризация.
Примером является реакция полимеризации стирола (винилбензола) с образованием полистирола: nCH2
= CH (C6
H5
) (- CH2
– CH (C6
H5
) -)n
Если мономеров два или более, имеет место сополимеризация:
nCH2
=CH – CH = CH2
+ nCH2
= CH – C =N → (- CH2
– CH = CH – CH2
– CH2
– CH (C=N) -)n
Например, при сополимеризации бутадиена и нитрила получают бутадиен-нитрильный каучук.
Многократно повторяющиеся в макромолекуле группы атомов – структурные звенья
– имеют тот же состав, что и мономер, но отличаются от него по строению. Число n называется степенью полимеризации
. Степень полимеризации для данного ВМС не является постоянной величиной, так как в макромолекулах может содержаться разное число молекул мономера. Обычно говорят о средней величине степени полимеризации и среднем значении молекулярной массы ВМС.
Реакция полимеризации протекает как самопроизвольный экзотермический процесс, но скорость ее без внешнего воздействия невелика. Полимеризация является цепной реакцией, она состоит из трѐх стадий: инициирования, наращивания цепи и обрыва цепи.
Полимеризация инициируется так называемыми свободными радикалами
– атомами или ионами, обладающими повышенной реакционной способностью за счет наличия неспаренного электрона. Чаще всего инициаторами полимеризации служат пероксиды водорода или бензоила. Активная частица (радикал) образует с молекулой мономера простую связь за счет одного электрона двойной связи. Другой электрон остается свободным, поэтому образовавшаяся частица также является радикалом, присоединяющим следующую молекулу мономера, и т.д. Процесс продолжается достаточно долго, пока велика концентрация мономера, а затем растущие радикалы могут встретиться и прореагировать друг с другом. Активные центры исчезнут, произойдет обрыв цепи.
При ионной полимеризации
роль активных центров играют катионы или анионы. Инициаторами катионной полимеризации служат кислоты (H2
SO4
, HCl,) и другие акцепторы электронов (SnCl4
, TiCl4
, AlCl3
), металлоорганические соединения типа Al(C2
H5
)3
и др. В качестве инициаторов анионной полимеризации служат электронодонорные вещества, в том числе щелочные металлы и их алкоголяты. Часто одновременно используют несколько инициаторов полимеризации для получения полимеров регулярной структуры.
Полимер называется стереорегулярным, если заместители R в основной цепи макромолекул (–CH2
–CHR–)n
расположены упорядоченно. Если все заместители находятся по одну сторону от плоскости цепи, то такие полимеры называют изотактическими (Рис. 6.2):
Рис. 6.2. Схема изотактического полимера

Если заместители располагаются в строгой очередности по одну и другую стороны от плоскости цепи, то такие полимеры называются синдиотактическими
(Рис. 6.3).
Рис. 6.3. Схема синдиотактического полимера

Стереорегулярные полимеры способны кристаллизоваться, они обладают большей прочностью и теплостойкостью.
Если боковые заместители в макромолекулах располагаются в беспорядке относительно плоскости основной цепи, то такой полимер является стереонерегулярным,
или атактическим
(Рис.6.4).
Рис. 6.4. Схема атактического полимера

Атактические полимеры не способны кристаллизоваться; по большинству эксплуатационных свойств они уступают стереорегулярным полимерам такого же химического состава.
Методом полимеризации производят более ¾ всего объема выпускаемых полимеров. Полимеризацию проводят в растворе, эмульсии, суспензии или газовой фазе.
Поликонденсацией
называется реакция получения из мономеров, содержащих две или более функциональные группы
, высокомолекулярных соединений с выделением побочных низкомолекулярных продуктов
(воды, аммиака, хлористого водорода и пр.). Это реакция замещения, при которой ступенчато образуются все более и более высокомолекулярные вещества. nC6
H5
OH + nHCHO [C6
H4
(OH) – CH2
- ]n
+ H2
O
– получение фенолформальдегидной смолы (фенопласта) из фенола и формальдегида. nHOOC – (CH2
)4
– COOH + nH2
N – (CH2
)6
–NH2

[- CO – (CH2
)4
–CO – NH – (CH2
)6
–NH -]n
+ nH2
O
- получение найлона-6,8 из адипиновой кислоты и гексаметилендиамина.
 Линейной называется поликонденсация соединений, содержащих две функциональные группы, например, аминокислот. Ниже приведена схема поликонденсации аминокапроновой
кислоты с получением капрона
(поли - - капроамида). Линейной называется поликонденсация соединений, содержащих две функциональные группы, например, аминокислот. Ниже приведена схема поликонденсации аминокапроновой
кислоты с получением капрона
(поли - - капроамида).
2 NH2
- (CH2
)5
– COOH NH2
– (CH2
)5
– CO – NH – (CH2
)5
– COOH + H2
O
NH2
– (CH2
)5
– CO – NH – (CH2
)5
– COOH + NH2
- (CH2
)5
– COOH
NH2
– (CH2
)5
– CO – NH – (CH2
)5
– CO – NH – (CH2
)5
– COOH + H2
O и т.д.
Поликонденсацию можно остановить на любой стадии.
Для ВМС, полученных путем поликонденсации, молекулы мономера и структурное звено отличаются по составу. Низкомолекулярные продукты реакции необходимо удалять из сферы реакции, так как их накопление приводит к росту скорости обратной реакции. Поликонденсацию ведут в расплаве или в растворе. Методом поликонденсации получают около ¼ выпускаемых полимеров.
Свойства полимеров зависят от строения их молекул.
Макромолекулы могут иметь различную форму:
1. Линейную, когда все структурные звенья последовательно соединены в одну линию (полиэтилен, полипропилен);
2. Разветвленную, когда у длинных цепей полимерных молекул имеются боковые ответвления (крахмал);
3. Пространственную, или сетчатую, когда линейные макромолекулы «сшиты» меж собой химическими связями (вулканизированный каучук – резина).
При нагревании
линейные полимеры размягчаются, плавятся, переходя в вязко-текучее состояние, а при дальнейшем нагревании разлагаются. Кристаллические полимеры, в отличие от аморфных, плавятся при строго определѐнной температуре. Пространственные полимеры разлагаются при нагревании, не расплавляясь. Термореактивные полимеры устойчивы к нагреванию. Все полимеры обладают высокой механической прочностью
вследствие сильного взаимодействия между отдельными макромолекулами, которое особенно сильно у пространственных полимеров, где макромолекулы связаны меж собой химическими связями. Механическую прочность ВМС можно увеличить путем добавления наполнителей, например, сажи и мела, армированием стекловолокном и т.п. Полимерные материалы, как правило, являются диэлектриками,
то есть не проводят электрический ток. Некоторые полимеры, имеющие сопряженные двойные связи (полиацетилен, поливинилены, полинитрилы) являются полупроводниками
. Полимеры могут подвергаться деструкции
под воздействием кислорода, света, теплоты и радиации. При этом уменьшается молекулярная масса полимеров, изменяются их физические и химические свойства. Старение полимеров
со временем замедляют введением стабилизаторов (фенолов, аминов и пр.)
Высокомолекулярные соединения применяются для получения композиционных материалов, или композитов
, когда полимерная основа укрепляется, или армируется прочными волокнами или кристаллами; пластмасс
для замены металла или дерева; текстильных волокон
; полимерных пленок
для магнитных носителей, изоляционных и упаковочных материалов; лаков, красок и клеев
.
6.4. Олигомеры
Отдельную группу составляют олигомеры, занимающие по размеру молекул область между мономерами и высокомолекулярными соединениями. Верхний предел молярных масс олигомеров зависит от их химической природы и соответствует тому значению, при котором начинают проявляться свойства, характерные для высокомолекулярных веществ. Молярные массы полярных олигомеров охватывают более широкий интервал (до ~1,5∙104
), чем неполярных (до ~5∙103
).
Большинство методов синтеза олигомеров основано на реакциях ограничения роста макромолекул в процессах полимеризации и поликонденсации. Кроме того, олигомеры получают деструкцией высокомолекулярных полимеров, а также ступенчатым синтезом с выделением продуктов реакции на каждой стадии. В последнем случае образуются монодисперсные олигомеры. Из множества реакционноспособных олигомеров можно выделить полиэфирные смолы, эпоксидные смолы, фенолоальдегидные смолы и прочие олигомеры, которые широко применяются в производстве слоистых пластиков, пенопластов, лаков, клеев, компаундов и т.д. Олигоолефины используют в качестве моторных топлив, смазочных масел, компонентов полировальных паст (синтетического воска), олигомеры фторзамещѐнных этилена — как высококипящие масла, теплоносители, жидкости для гидроприводов. Олигомеры на основе окисей олефинов широко применяются как синтетические поверхностно-активные вещества (СПАВ).
6.5. Вопросы и задания:
1. Чем определяется громадное разнообразие органических молекул?
2. Что такое изомерия? Какие виды изомерии вы знаете?
3. Перечислите основные функциональные группы и соответствующие им классы органических соединений.
4. Дайте характеристику природным полимерам.
5. На какие группы делятся синтетические ВМС?
6. Какие способы получения полимерных соединений (ВМС) вы знаете?
7. Чему равна степень полимеризации бутилена СН=СН-СН-СН при получении изобутилена с Мч
= 56000?
8. Составьте уравнение полимеризации ацетилена С2
Н2
. Какими специфическими свойствами обладает полиацетилен?
9. Составьте уравнение получения найлона-6,8 при поликонденсации гексаметилендиамина и адипиновой кислоты. Какое количество мономеров следует взять, чтобы получить ВМС со степенью полимеризации 1000?
10. Волокно энант получают поликонденсацией аминоэнантовой кислоты H2
N – (CH2
)6
–COOH. Напишите уравнение поликонденсации и определите массу аминоэнантовой кислоты, которая потребуется для получения 150 г волокна.
Лекция 7. Химическая термодинамика и кинетика
7.1. Химическая термодинамика
Термодинамика
– это наука о взаимных превращениях различных форм энергии: тепловой, электрической, химической, механической и т.д. Таким образом, современная термодинамика охватывает все процессы, с которыми сталкивается человек в своей практической деятельности.
Термодинамической системой
называется любая совокупность частиц или физических тел, которые каким-либо образом взаимодействуют между собой. Системой может быть любая исследуемая макроскопическая
часть Вселенной. Обычно это совокупность тел, условно отделенных от окружающей среды. Например, раствор в колбе, сжатый поршнем газ, тепловая машина, космический корабль и т.д. Окружающей средой
называется все находящееся за пределами системы. Термодинамическая система, лишенная возможности обмена с окружающей средой как массой, так и энергией, называется изолированной
. В природе такие системы не существуют, они имеют лишь теоретическое значение. Термодинамическая система, соприкасаясь с окружающей средой, может вступать с ней в обмен и веществом, и энергией. Если это происходит, то система называется открытой
. Термодинамическая система, лишенная возможности обмениваться веществом с окружающей средой, но обменивающаяся с ней энергией, называется закрытой
.
Состояние
термодинамической системы определяется ее термодинамическими параметрами, или функциями состояния
. Функцией состояния
называется такая характеристика системы, изменение которой при переходе системы из одного состояния в другое не зависит от вида соответствующего этому переходу термодинамического процесса, а целиком определяется значениями параметров начального и конечного состояний. Для химической системы такими параметрами могут быть масса, давление, объем, температура и концентрация реакционной смеси. Эти параметры, или функции состояния
термодинамической системы, являются интенсивными величинами
, так как не зависят от массы системы. Экстенсивные величины
зависят от массы системы. Это, например, внутренняя энергия термодинамической системы,
которая представляет собой сумму энергий движения и взаимодействия всевозможных частиц, их которых состоит система. Экстенсивными величинами являются такие функции состояния системы, как энтальпия
, энтропия
и химический потенциал
.
Химическая термодинамика изучает процессы, происходящие в химических системах. Как и все остальные системы Вселенной, химические системы подчиняются законам термодинамики.
7.2. Законы термодинамики
Первый закон термодинамики
, или закон сохранения энергии, сформулированный в 1847 г. немецким физиком Германом Людвигом Фердинандом Гельмгольцем гласит:
Мир состоит из атомов, атомы обладают потенциальной (тяготение) и кинетической (движение) энергией. Сумма потенциальных и кинетических энергий частиц, из которых построено тело или система тел, не может измениться, если эта система не подвержена внешним воздействиям.
Другими словами, полная энергия изолированной системы есть величина постоянная.
Закон сохранения энергии – это строжайший «бухгалтер» прихода и расхода энергии. В любом явлении приход должен точно сойтись с расходом. Энергия может переходить из одной формы в другую, но не может возникать или исчезать. Современная физика знает, что энергия и масса эквивалентны и связаны меж собой знаменитым эйнштейновским соотношением Е=mc2
, где Е – энергия системы; m – еѐ масса, а c – скорость света. Это значит, что масса может превращаться в энергию, а энергия – рождать массу. Однако на макроуровне, в частности, в химических системах, масса превращается в энергию в такой незначительной степени, что закон сохранения массы и закон сохранения энергии можно считать независимыми законами. Первый закон термодинамики не ограничивает протекание термодинамических процессов.
Ограничение на них накладывает второй закон термодинамики
. Этот закон был сформулирован в 1850 году немецким физиком Рудольфом Клаузиусом (Готлибом) в таком виде: «самопроизвольная передача тепла от менее нагретого тела к более нагретому невозможна».
В 1850 Клаузиус ввел понятие энтропии как свойства системы в процессе работы превращать часть своей внутренней энергии на бесполезную теплоту.
Второй закон термодинамики можно назвать «налоговым инспектором», взимающим налог в виде энтропийной теплоты за любую произведенную системой работу. В изолированной системе энтропия стремится к максимуму
. Энтропия есть мера хаоса,
так как чем больше в системе энтропийной теплоты, тем быстрее хаотической движение составляющих ее молекул, тем больше беспорядок в данной системе. Это значит, что второй закон термодинамики носит вероятностный характер: система самопроизвольно стремится в состояние большего хаоса потому, что такое состояние более вероятно, чем порядок.
В изолированной системе энтропия стремится к максимуму.
Австрийский учѐный Людвиг Больцман (1844-1906) нашел связь между законом возрастания энтропии и случайностью. Математически она имеет вид:
SK
lgW
, (7.1)
где S - энтропия, К – константа Больцмана, равная 1,38∙10-23 Дж/град, W – число возможных микросостояний частиц, составляющих макросистему, посредством которых может реализоваться макросостояние системы; W называют также термодинамической вероятностью
макросостояния системы.
Рассмотрим воображаемую систему из двух сосудов с одинаковым газом, разделенных заслонкой. Предположим, что мы сначала нагрели газ в левом сосуде, а затем открыли заслонку. Через некоторое время температура в обоих сосудах выровняется и наступит состояние равновесия: параметр «температура» будет оставаться постоянным, так как мы считаем свою систему изолированной, лишенной обмена энергией с окружающей средой. Равновесие наступило потому, что более «горячие» частицы, сталкиваясь с более «холодными», сообщили им часть своей энергии. В результате произошло выравнивание энергии между всеми частицами. А можно ли совершить обратный процесс и направить все «горячие» молекулы в левый сосуд, а «холодные» оставить в правом? Английский естествоиспытатель Джеймс Клерк Максвелл (1831 -1879), занимаясь изучением газовых систем, задался тем же вопросом. Для того, чтобы получить в левом сосуде газ с более высокой температурой без нагревания, то есть систему с заданным порядком – горячие молекулы слева, холодные справа - Максвелл «посадил» у перегородки некоего «любителя порядка», – физики называют его «демоном Максвелла», – который должен бы был сортировать хаотически перемешанные молекулы газа, перенося только «горячие», то есть имеющие большую скорость, молекулы налево, а
«холодные» с меньшими скоростями направо. Сколькими способами можно получить желаемый порядок? Только одним
, когда у перегородки будет сидеть «демон Максвелла» и направлять молекулы туда, куда следует. А сколькими способами можно получить состояние, отличное от «заказанного» порядка? Первое такое состояние наступит, когда одна «горячая» молекула останется в правом сосуде, а одна «холодная» окажется в левом. Если учесть, что в 1 кубическом сантиметре воздуха находится 2,687∙1019
молекул (число Лошмидта), количество возможных «беспорядочных» микросостояний достигает 10300000000000000000000
. Понятно, что вероятность перехода в состояние беспорядка неизмеримо выше, и именно такой переход и происходит в изолированной системе самопроизвольно
, то есть без вмешательства извне. Чисто теоретически самопроизвольный переход системы в упорядоченное состояние возможен, но такое поведение системы крайне маловероятно. Из (7.1) вытекает, что энтропия S=0 в той системе, для которой W=1. Исходя из этого, в 1911 году Макс Планк предложил следующий постулат: энтропия идеального кристалла чистого вещества при абсолютном нуле равна нулю
. Этот постулат называют третьим (или нулевым) законом термодинамики. Абсолютным нулѐм
называется такая температура, при которой отсутствует любое движение составляющих систему элементов, или W = 1. Это ноль градусов по шкале Кельвина, или -273 градуса по шкале Цельсия. На основании третьего закона термодинамики можно определить значения энтропии системы в произвольном равновесном состоянии.
Повышение температуры любого тела ведет к увеличению энтропии. В фазовых переходах (плавление, кристаллизация, кипение, конденсация, сублимация и т.п.) энтропия меняется скачкообразно при постоянной температуре.
7.3. Химическая кинетика
Химическая кинетика изучает факторы, влияющие на скорость
процессов, протекающих в химических системах, а также механизмы
этих процессов.
Различные реакции в химических системах могут протекать с разными скоростями. Например, куча угля может годами лежать возле котельной, практически не уменьшаясь в объеме, так как химическая реакция: С + О2
= СО2
протекает крайне медленно. А вот реакция нейтрализации кислоты щелочью: НCl + NaОН = Н2
О + NaCl
протекает практически мгновенно.
При рассмотрении вопроса о скорости реакции необходимо различать реакции, протекающие в гомогенной системе (гомогенные реакции
), и реакции, протекающие в гетерогенной системе (гетерогенные реакции
). Гомогенной называется система реагирующих веществ, состоящая из одной фазы
. Скоростью гомогенной реакции
называется изменение концентрации реагирующих веществ в единицу времени. Скоростью гетерогенной реакции
называется изменение поверхностной концентрации реагирующих веществ в единицу времени. Поверхностной концентрацией называется концентрация реагирующих веществ, приходящаяся на единицу реакционной поверхности.
Все вещества построены из молекул, атомов или ионов, поэтому для того, чтобы вступить в реакцию, две реагирующие частицы должны сблизиться друг с другом и столкнуться. Такая модель называется моделью активных соударений
. Скорость химической реакции будет тем выше, чем больше реагирующих частиц сталкивается друг с другом. Число соударений будет, в свою очередь тем выше, чем выше концентрация исходных веществ, или их произведение концентраций:
Для реакции общего вида: xА + yB = nC + mD v = k [A]x
[B]y
(7.2)
Зависимость (7.2) называется кинетическим уравнением
, или законом действующих масс,
который норвежские учѐные К. Гульдберг и П. Вааге открыли в 1864-1867 годах. В соответствии с законом действующих масс, скорость химической реакции равна произведению молярных концентраций реагирующих веществ, взятых в степенях, равных коэффициентам, стоящим перед данными веществами в уравнении соответствующей реакции, с учѐтом константы скорости реакции.
Константа скорости реакции k
зависит только от природы реагирующих веществ.
Интересен вопрос о том, почему в кинетическом уравнении (7.2.) фигурирует именно произведение
молярных концентраций реагентов, а, например, не сумма? Для того чтобы столкнуться, молекулы А
и B
должны оказаться одновременно в какой-то точке пространства. Вероятность того, что два независимых события произойдут одновременно (молекулы А
и B
окажутся в одно время в одном и том же месте) равна произведению вероятностей
каждого из этих событий по отдельности. Это положение теории вероятностей легко проверяется. Наибольшую вероятность обозначают единицей. Например, вероятность того, что подброшенная вверх монета упадет плашмя, практически равна 1. Вероятность того, что монета упадет орлом вверх, равна 1/2. Если мы подбросим одновременно две монеты, то вероятность того, что обе они упадут орлом вверх, составляет 1/2.
1/2 = 1/4. Это означает, что в серии из 4-х опытов с подбрасыванием монет только один раз выпадут два орла. Если в маленькой серии опытов и произойдет отклонение от теории, то в большой серии (например, из 100 опытов), таких отклонений уже практически не наблюдается. Вероятность для молекул А одновременно оказаться в одном и том же месте прямо пропорциональна молярной концентрации этих молекул
[А]. Это же можно сказать о молекулах B. Следовательно, вероятность
их столкновения должна быть пропорциональна произведению
молярных концентраций [А]∙[Б].
Если все компоненты системы, в которой протекает химическая реакция, находятся в газовой фазе, то вместо концентрации можно пользоваться параметрами давления. Например, для гомогенной реакции окисления моноксида азота (II): 2NO + O2
= 2NO2
;
кинетическое уравнение можно записать либо в виде:
v = k∙[NO]2
[O2
] (7.3),
где [NO] и [O2
] – молярные концентрации моноксида азота и кислорода
соответственно,
либо в виде:
 V kP
NO
2
P
O
2
,
(7.4), V kP
NO
2
P
O
2
,
(7.4),
где PNO
и PO2
-
парциальные давления моноксида азота и кислорода
соответственно.
В случае гетерогенных процессов в уравнение закона действующих масс входят только концентрации веществ, находящихся в газовой фазе или в растворе. Концентрация веществ, находящихся в твердой фазе, считается постоянной величиной и входит в константу скорости реакции.
Например, для реакции горения угля:
С + О2
= СО2
закон действия масс запишется так:
v = k1
∙ const ∙ [О2
] = k[О2
] ;
Факторы, определяющие скорость реакции, будут подробно рассмотрены в Лекции 10.
Если уравнение реакции сложное, то, скорее всего, реакция включает в себя несколько более простых реакций, каждая из которых происходит путем попарных столкновений, либо путем распада одной частицы. Эти простые процессы называют элементарными
реакциями.
Кинетическое уравнение (7.2) справедливо только для таких элементарных реакций.
Например, для реакции окисления бромистого водорода кислородом воздуха, протекающей достаточно медленно:
4HBr(г) + O2(г) → 2H2O(г) + 2Br2(г) (7.5)
кинетическое уравнение имеет вид
v
= k[HBr]4
[O2
] (7.6)
В прямой реакции должны участвовать одновременно 5 молекул, но вероятность такого столкновения в газовой фазе практически равна нулю. Такая сложная реакция на самом деле включает в себя несколько простых (элементарных) реакций, каждая из которых описывается своим собственным (простым) кинетическим уравнением и имеет свою константу скорости. В итоге общую скорость реакции определяет какаято самая медленная
элементарная реакция. Такая реакция называется лимитирующей стадией
.
Общее кинетическое уравнение для химической реакции можно найти только экспериментально. В данном случае из опыта следует, что изменение концентрации (или давления) HBr оказывает точно такое же влияние на скорость реакции данной реакции, как и изменение концентрации (давления) О2,
хотя из кинетического уравнения (7.6) следует, что изменение концентрации HBr должно влиять на скорость реакции в гораздо большей степени ([HBr]4
). Объясняется это тем, что данная реакция протекает в три стадии. Если написать уравнение реакции, включающее только исходные вещества и конечные продукты, но и все промежуточные вещества в этой реакции (часто они неустойчивы, и выделить их невозможно), то получится запись механизма реакции
:
(1) HBr(г)
+ O2(г)
→ НООBr(г)
(медленно)
(2) НООBr(г)
+ HBr(г)
→ 2НОBr(г)
(быстро)
(3) НОBr(г)
+ HBr(г)
→ H2
O(г)
+ Br2(г)
(быстро)
Лимитирующей стадией суммарной реакции (7.5) является самая медленная стадия (1). Именно поэтому изменение концентраций HBr и О2
оказывает одинаковое воздействие на скорость реакции. Знание механизмов реакций позволяет управлять ходом химических процессов, хотя для выяснения каждого механизма необходимы серьѐзные дополнительные исследования.
Величины показателей степени при концентрациях реагентов называют порядком реакции
по данному веществу. Молекулярность
реакции определяется числом молекул, одновременным взаимодействием которых осуществляется элементарный акт химического превращения. Например, реакция разложения молекулярного йода
I2
= 2I
является мономолекулярной; реакция разложения йодида водорода
2HI = H2
+ I2
бимолекулярная. Определить порядок и молекулярность реакции не всегда просто, так как часто уравнение реакции не отражает ее механизма. Кинетическое уравнение общего вида v
= k[A]a
[B]b
[C]c
имеет порядок
по каждому из входящих в него веществ. Порядок реакции
по данному веществу - это показатель степени при концентрации данного вещества в кинетическом уравнении. Например, кинетическое уравнение v
= k[NO]2
[O2
] имеет второй порядок по оксиду азота NO и первый порядок по кислороду О2
. Сумма порядков по всем веществам называется общим
или суммарным порядком реакции
. Например, кинетическое уравнение v
= k[H+
][OH-
] имеет общий второй
порядок. Уравнение v
= k[NO]2
[O2
] имеет общий третий
порядок. Уравнение v
= k[K2
Cr2
O7
] - первого
порядка. В реальных кинетических процессах редко встречается порядок реакции выше третьего, то есть ни для одной из стадий механизма реакции не требуется одновременного столкновения более чем двух частиц
.
7.4. Вопросы и задания
1. Что такое термодинамическая система?
2. Сформулируйте первый, второй и третий закон термодинамики.
3. Что такое энтропия?
4. Сколько микросостояний имеет система, состоящая из 4 молекул, две из которых «горячие», а две – «холодные»?
5. Что такое «абсолютный нуль»?
6. Как изменяется энтропия в следующих процессах?
H2
O(ж)→H2
O(г); J2
(к)→J2
(г); 4NH3
(г)+3O2
(г)→6H2
O(г)+2N2
(г);
CO(г)+3H2
(г)→CH4
(г)+H2
O(г)
7. Что такое скорость гомогенной химической реакции? Гетерогенной химической реакции?
8. Сформулируйте закон действующих масс.
9. Что такое порядок реакции?
10. Почему в реальных условиях практически не встречаются реакции с порядком выше третьего?
Лекция 8. Энергетика химических процессов
Химические системы, в которых происходит превращение энергии, подчиняются термодинамическим законам. Химические реакции часто протекают с выделением или поглощением энергии. Обычно эта энергия выделяется или поглощается в виде тепла (реже в виде света или механической энергии). Например, процесс горения метана, протекающий по схеме CH4
+ 2O2
= CO2
+ H2
O
сопровождается значительным выделением тепловой энергии; наоборот, реакция образования оксида азота (II) по схеме: N2
+ O2
= 2NO
требует подвода теплоты извне и не идет при отсутствии нагревания реакционной смеси.
Реакции, протекающие с выделением тепловой энергии, называются экзотермическими
(от латинского слова экзо
– наружу), а реакции, в которых энергия поглощается, называются эндотермическими
(от латинского слова эндо
– внутрь).
8.1. Термохимия
Количество теплоты, выделяющееся или поглощающееся при реакции, называется тепловым эффектом и обозначается как
H
. Раздел химии, посвященный изучению тепловых эффектов химических систем, называется термохимией.
Теплоту можно измерить в специальных приборах – колориметрах. Величину теплового эффекта химической реакции выражают в джоулях на один моль
какого-либо исходного вещества (реже – продукта реакции). Поэтому в термохимических уравнениях
коэффициенты могут быть дробными. Например, термохимическое уравнение сгорания водорода в кислороде с образованием воды выглядит следующим образом:
 Н2
(г)+ 1/2О2
(г) = Н2
О(ж); H
– 285,84 кДж/моль (8.1) Н2
(г)+ 1/2О2
(г) = Н2
О(ж); H
– 285,84 кДж/моль (8.1)
Принято также указывать агрегатное состояние участвующих в реакции веществ; ясно, что тепловой эффект образования водяного пара превышал бы тепловой эффект образования жидкой воды на теплоту, необходимую для осуществления фазового перехода Н2
О(ж) → Н2
О(г). Кроме того, в соответствии с рекомендациями Международного союза по теоретической и прикладной химии (ИЮПАК), теплота, поступление которой извне требуется химической системе на осуществление реакции, считается положительной, а теплота, которая выделяется из системы в результате реакции – отрицательной
. Реакция (8.1.) протекает с выделением теплоты (в системе осуществляется экзотермический процесс), поэтому перед значением теплового эффекта стоит знак минус.
Откуда же берется тепло при химической реакции, или на что оно расходуется? Рассмотрим в качестве примера реакцию горения метана, которая идет по схеме:
CH4
(г)+ 2O2
(г) = CO2
(г) + H2
O (ж) (8.2)
Для того, чтобы перейти от состояния «исходные вещества» к состоянию «продукты реакции», нужно разорвать четыре связи С-Н и две связи С=О, а из возникших при этом атомов С, Н и О «собрать» молекулу О=С=О и две молекулы Н-О-Н. Разрыв устойчивых химических связей всегда требует затраты энергии, равной сумме энергий разрушаемых связей. Соединение атомов в молекулы, то есть образование химических связей, сопровождается выделением энергии, равной сумме энергий образовавшихся связей. Теплота при горении метана выделяется потому, что сумма энергий связей в молекулах продуктов СО2
и 2Н2
О больше суммы энергий связей в молекулах СН4
и 2О2
. Или, другими словами, на разрыв связей в исходных веществах требуется меньше энергии, чем еѐ выделяется при образовании продуктов реакции. «Избыток» энергии выделяется из системы при горении метана в виде 890,2 кДж теплоты на один моль метана. Если мы захотим провести обратную реакцию получения метана и кислорода из диоксида азота и воды, то нам придѐтся затратить те же 890,2 кДж/моль теплоты.
Такое заключение вытекает из первого закона термохимии
, или закона Лавуазье–Лапласа
(который является следствием первого закона термодинамики):
Тепловой эффект прямой реакции всегда равен тепловому эффекту обратной реакции с противоположным знаком.
Это означает, что при образовании любого соединения выделяется (поглощается) столько же энергии, сколько поглощается (выделяется) при его распаде на исходные вещества. Например, горение водорода в кислороде с выделением теплоты:
 Н2
(г)+ 1/2О2
(г) → Н2
О(ж);
H
– 285,84 кДж/моль
можно считать прямым процессом. При этом образуются на одну молекулу воды две водород-кислородные связи. Тогда обратным процессом будет разложение воды на водород и кислород, и на разложение одного моля воды придется затратить такое же количество энергии (например, электрической) Н2
(г)+ 1/2О2
(г) → Н2
О(ж);
H
– 285,84 кДж/моль
можно считать прямым процессом. При этом образуются на одну молекулу воды две водород-кислородные связи. Тогда обратным процессом будет разложение воды на водород и кислород, и на разложение одного моля воды придется затратить такое же количество энергии (например, электрической)
H2
О(ж) → H2
(г) + O2
(г) + 285, 84 кДж/моль,
так как для разложения воды придѐтся разорвать водород-
кислородные связи, энергия которых остаѐтся постоянной.
Задача термохимии состоит в том, чтобы посредством предварительных расчѐтов предсказать, каковы будут тепловые эффекты тех или иных реакций. В химической промышленности тепловые эффекты нужны для расчета количества теплоты для нагревания реакторов, в которых идут эндотермические реакции, или для того, чтобы обеспечить надлежащее охлаждение реакторам, в которых идут экзотермические процессы. В теплоэнергетике с помощью тепловых эффектов сгорания различных видов топлива рассчитывают выработку тепловой энергии. Врачи-диетологи используют тепловые эффекты окисления пищевых продуктов в организме человека для составления правильных рационов питания. Расчет теплового эффекта реакции по энергии химических связей возможен, но применяется нечасто, так как измерять эту энергию, особенно в молекулах с большим числом связей, сложно и дорого. Упростить расчѐты энергетики реакций помогает второй закон термохимии, который был сформулирован в 1840 г российским академиком Г. И. Гессом:
Тепловой эффект реакции зависит только от начального и конечного состояния веществ и не зависит от промежуточных стадий процесса.
Это означает, что общий тепловой эффект ряда последовательных реакций будет таким же, как и у любого другого ряда реакций, если в начале и в конце этих рядов находятся одни и те же исходные и конечные вещества
. Это значит, что законы термохимии придают термохимическим уравнениям некоторое сходство с математическими функциями; это значит, в термохимических уравнениях можно переносить члены из одной части в другую, почленно складывать, вычитать и сокращать формулы химических соединений. При этом необходимо учитывать коэффициенты в уравнениях реакций и не забывать о том, что складываемые, вычитаемые или сокращаемые моли вещества должны находиться в одинаковом агрегатном состоянии.
Предположим, что нам известны тепловые эффекты следующих реакций:
 ½ N2
(г) + ½ O2
(г) → NO (г); H
+90,72 кДж/моль (8.3) ½ N2
(г) + ½ O2
(г) → NO (г); H
+90,72 кДж/моль (8.3)
½ N2
(г) + O2
(г) → NO2
(г);
H
+34,02 кДж/моль
(8.4) Требуется определить тепловой эффект следующего процесса:
 NO (г) + ½ O2
(г) → NO2
(г); H
? (8.5) NO (г) + ½ O2
(г) → NO2
(г); H
? (8.5)
Для этого нам нужно вывести уравнение реакции (8.5) из уравнений (8.3) и (8.4). Поскольку моноксид азота NO в уравнении реакции (8.5) является исходным веществом, нам нужна реакция, обратная реакции
(8.3):
NO (г) → ½ O2
(г) + ½ N2
(г);
H
-90,72 кДж/моль
(8.6) По закону Лавуазье-Лапласа знак теплового эффекта при этом меняется на противоположный.
Складываем уравнения (8.6) и (8.4) и, соответственно, их тепловые эффекты (составляем цикл Гесса
):
 NO (г) → ½ O2
(г) + ½ N2
(г);
H
-90,72 кДж/моль NO (г) → ½ O2
(г) + ½ N2
(г);
H
-90,72 кДж/моль
+
 ½ N2
(г) + O2
(г) → NO2
(г);
H
9.4
+34,02 кДж/моль ____________________________________________________________ NO (г) + ½ N2
(г) + O2
(г) → ½ O2
(г) + ½ N2
(г) + NO2
(г);
H
-90,72 кДж/моль + 34,02 кДж/моль; ½ N2
(г) + O2
(г) → NO2
(г);
H
9.4
+34,02 кДж/моль ____________________________________________________________ NO (г) + ½ N2
(г) + O2
(г) → ½ O2
(г) + ½ N2
(г) + NO2
(г);
H
-90,72 кДж/моль + 34,02 кДж/моль;
сокращаем подобные члены и получаем:
 NO (г) + ½ O2
(г) → NO2
(г); H
-56,7 кДж/моль NO (г) + ½ O2
(г) → NO2
(г); H
-56,7 кДж/моль
Составлять циклы Гесса, подбирая и преобразовывая реакции с известными тепловыми эффектами, далеко не всегда просто. Поэтому существует другой способ расчѐта тепловых эффектов. Один моль любого вещества обладает определѐнным теплосодержанием
, которое является мерой энергии, которую вещество накапливает при образовании. Теплосодержание можно выразить через теплоту образования этого вещества из простых веществ
.
8.2 Теплота образования
Теплотой образования
данного соединения называется количество теплоты, выделяющееся при образовании 1 моль этого соединения из простых веществ.
Например, выражение «теплота образования жидкой воды равна - 285,8 кДж/моль» означает, что при образовании 1 моль (18 г) жидкой воды из 1 моль (2 г) водорода и ½ моль (16 г) кислорода выделяется именно такое количество теплоты. Смешав в калориметре весовые количества водорода и кислорода, точно соответствующие 1 моль водорода и ½ моль кислорода, можно измерить теплоту образования одного моль воды при взрыве гремучего газа (- 285,8 кДж/моль). Подобным же образом можно измерить теплоту образования других веществ, например, входящих в уравнение реакции (8.2). Так, теплота образования метана равна -74,9 кДж/моль, а теплота образования диоксида углерода равна -393,5 кДж/моль. Кислород является простым веществом, а теплоты образования всех простых веществ принимаются равными нулю
. При этом имеется в виду наиболее распространѐнная в данных условиях форма простого вещества, с соблюдением предварительных договорѐнностей. Так, например, при термохимических измерениях стандартным состоянием углерода считается графит, а не алмаз или уголь.
 Для удобства сравнения теплот образования их рассчитывают для случая, когда температура исходных веществ и продуктов реакции равна 250
С, или 298,16 К по абсолютной шкале. Теплоты образования, относящиеся к этой температуре, обозначают H
0
обр
.298
и называют стандартными при температуре 298,16К. Стандартная теплота образования вещества равна теплоте реакции образования 1 моль этого вещества из простых веществ в стандартных условиях. Для удобства сравнения теплот образования их рассчитывают для случая, когда температура исходных веществ и продуктов реакции равна 250
С, или 298,16 К по абсолютной шкале. Теплоты образования, относящиеся к этой температуре, обозначают H
0
обр
.298
и называют стандартными при температуре 298,16К. Стандартная теплота образования вещества равна теплоте реакции образования 1 моль этого вещества из простых веществ в стандартных условиях.
Решением Международного союза теоретической и прикладной химии (ИЮПАК
) в 1975 году стандартное состояние определено следующим образом: «Стандартным состоянием для газов является состояние гипотетического идеального газа при давлении в 1 физическую атмосферу (101325 Па). Для жидкостей и твердых веществ стандартным состоянием является состояние чистой жидкости или соответственно чистого кристаллического вещества при давлении в 1 физическую атмосферу. Для веществ в растворах за стандартное состояние принято гипотетическое состояние, при котором энтальпия одномолярного раствора (1 моль вещества в 1 кг растворителя) равнялась бы энтальпии раствора при бесконечном разбавлении. Свойства веществ в стандартных состояниях обозначаются надстрочным индексом 0». Чистым веществом называется вещество, состоящее из одинаковых структурных частиц (атомов, молекул и др.).
Тепловой эффект реакции взаимодействия, например, водорода и кислорода с образованием воды (8.1) мы назовем стандартной теплотой образования
воды из простых веществ только при условии, что изначально водород и кислород находились при атмосферном давлении и температуре 298 К, после чего произошла реакция с выделением большого количества теплоты, а затем продукт реакции (вода) вновь был охлажден до 298 К, отдав все полученное во время реакции тепло в окружающую среду. Стандартная теплота образования жидкой воды составляет -285,84 кДж/моль.
8.3 Энтальпия
Теплота образования химического соединения, находящегося в стандартном состоянии, из простых веществ, также находящихся в стандартном состоянии и в устойчивых при данной температуре и давлении фазах и модификациях, называется также стандартной энтальпией
образования химического соединения. Энтальпия характеризует теплосодержание вещества и является функцией состояния термодинамической системы.
Зачем понадобилось вводить такое понятие, как энтальпия, которую обозначают таким же символом H
, как и тепловой эффект, и чем они отличаются друг от друга? Дело в том, что наиболее удобным методом измерения тепловых эффектов для химиков долгое время служил способ проведения реакций в «бомбе» - замкнутом металлическом сосуде, который помещают в калориметр. В замкнутом сосуде продукты реакции лишены возможности изменять объем, поэтому они не могут выполнить какую-нибудь механическую работу. В этих условиях выделившееся сквозь стенки «бомбы» теплота условно равна внутренней энергии системы U
, хотя это еще не вся энергия, заключавшаяся в данной реакции. Если давление в реакции возрастает, а «бомба» окажется не очень прочной, то еѐ просто разорвет, причем на эту работу будет потрачено еще какое-то количество энергии, которое мы «не замечаем» в том случае, если «бомба» осталась цела. Но большинство химических реакций химики проводят не при постоянном объеме (не в «бомбе»), а в открытых сосудах (т.е. при постоянном давлении). Поэтому потребовалась величина, аналогичная U
, но измеряемая для реакций в открытых сосудах. Именно эта величина и называется энтальпией. Это тепловой эффект реакции, измеренный (или вычисленный) для случая, когда реакция происходит в открытом сосуде (т.е. при неизменном давлении, или в изобарноизотермическом процессе).
Когда объем, занимаемый продуктами реакции, отличается от объема, занимаемого реагентами, химическая система может совершить дополнительную работу P V
(где P - давление, V
- изменение объема). Поэтому энтальпия H
и внутренняя энергия U
связаны между собой соотношением:
 H U P V
. H U P V
.
Это значит, что если реакция проводится не в «бомбе», то понятия «энтальпия» и «тепловой эффект» совпадают
. Если мы проводим реакцию получения воды в открытом сосуде, то 285,84 кДж/моль - это тепло, содержащееся в водороде и кислороде для случая, когда мы получаем из них воду.
Если при измерении теплот образования вещества, участвующие в реакции горения метана (8.2), находились в стандартных условиях (при температуре 298 К и давлении 1 атмосфера, или 101 кПа), то их теплоты образования являются стандартными теплотами образования
, или, что то же самое, стандартными энтальпиями образования
, и обозначаются, соответственно, как H
0
обр
.298
.
Существуют обширные таблицы стандартных энтальпий образования
. Эти данные очень полезны для расчѐта тепловых эффектов реакций.
 Для удобства сравнения тепловых эффектов их также рассчитывают для случая, когда температура исходных веществ и продуктов реакции равна 250
С, или 298,16 К. Тепловые эффекты, относящиеся к этой температуре, обозначают H
298
0
и называют стандартными тепловыми эффектами при температуре 298,16К. Для удобства сравнения тепловых эффектов их также рассчитывают для случая, когда температура исходных веществ и продуктов реакции равна 250
С, или 298,16 К. Тепловые эффекты, относящиеся к этой температуре, обозначают H
298
0
и называют стандартными тепловыми эффектами при температуре 298,16К.
При постоянной температуре и давлении
(в изобарноизотермическом процессе) тепловой эффект реакции равен изменению энтальпии процесса, которое также обозначается
H
298
0
. В химических процессах энтальпия H
связана с изменением внутренней энергии системы U
соотношением:
 U H nRT
, (8.7) U H nRT
, (8.7)
Где Δn – изменение числа моль газообразных продуктов реакции и исходных веществ, R - универсальная газовая постоянная, равная 8,3144 Дж/моль∙К, T – температура в градусах Кельвина.
Вычислим тепловой эффект (энтальпию)[11]
сгорания метана, исходя из стандартных теплот (энтальпий)[12]
образования метана (-74,9 кДж/моль) и продуктов его сгорания – жидкой воды (-285,8 кДж/моль) и диоксида углерода (-393,5 кДж/моль). Стандартная энтальпия кислорода как простого вещества равна нулю.
Для того чтобы реакция горения метана (8.2): CH4
(г)+ 2O2
(г) = CO2
(г) + 2 H2
O (ж)
осуществилась,
нужно разложить 1 моль метана (то есть затратить теплоту, равную теплоте образования метана H
0
обр
.298
с обратным знаком) и позволить образоваться 1 моль диоксида азота (при этом выделится H
0
обр
.298
диоксида углерода) и 2 моль воды (при этом выделится 2 H
0
обр
.298
воды). Это значит, что для расчѐта энтальпии данной реакции необходимо сложить энтальпии образования продуктов реакции (диоксида углерода и воды, с учѐтом количества моль каждого вещества) и вычесть из этой суммы сумму энтальпий образования исходных веществ (в данном случае это только метан, так как энтальпия образования кислорода равна нулю).
Сделанный нами вывод есть следствие из закона Гесса
, которое лежит в основе расчѐта энтальпий химических реакций на основе энтальпий образования сложных веществ. Оно гласит:
Энтальпия химической реакции равна сумме энтальпий образования продуктов реакции за вычетом суммы энтальпий образования исходных веществ, с учѐтом количества моль каждого вещества.
Математически его можно записать следующим образом:
 H х
.р
. H
прод
(8.8) H х
.р
. H
прод
(8.8)
Для реакции (8.2) это выражение будет иметь вид:
H
0 х
. o o
 CHo
4}, или CHo
4}, или
Hх
.р
. = {2∙ (-285,8) + (-393,5) – 0 –( -74,9 ) } = -890,2 кДж/моль.
8.4 Теплота сгорания
Законы термохимии применимы ко всем процессам, протекающим с поглощением или выделением теплоты, однако особое значение имеют реакции сгорания различных видов топлива. Теплоты сгорания достаточно легко измерить на практике и применить для расчѐта энтальпий различных процессов, а также для определения энтальпий образования сложных веществ, которые нелегко, а иногда и невозможно, получить из простых веществ.
Пусть, например, требуется вычислить энтальпию образования бензола C6
H6(ж)
из простых веществ, если известна его теплота сгорания при Т= 298К и Р=1,0133∙10-5
Па (-3267,7 кДж/моль), продуктами сгорания являются диоксид углерода CO2(г)
и вода H2
O(ж)
, а теплоты сгорания углерода С и водорода Н2
соответственно равны:

С(графит)
+ О2
(г) = СО2(г)
;
– 393,51 кДж/моль
 Н2 (г)
+ 1/2О2 (г)
= Н2
О(ж)
;
H – 285,84 кДж/моль. Н2 (г)
+ 1/2О2 (г)
= Н2
О(ж)
;
H – 285,84 кДж/моль.
Образование бензола из простых веществ осуществляется по уравнению:
6С (графит)
+ 3Н2(г)
= C6
H6(ж)
;
H
0
=?
(8.9)
бр
Составим следующий цикл Гесса:
6С(графит)
+ 6О2(г)
= 6СО2(г)
;
Нх
.р
.= 6∙(-393,51) кДж/моль
(8.10) 3Н2 (г)
+ 3/2О2 (г)
= 3Н2
О(ж)
;
Нх
.р
. = 3∙ (-285,84) кДж/моль
(8.11)
 C6
H6(ж)
+ 15
О2(г)
= 6CO2
(г)
+ 3H2
O(ж)
; Нх
.р
. = -3267,7 кДж/моль (8.12) 2 C6
H6(ж)
+ 15
О2(г)
= 6CO2
(г)
+ 3H2
O(ж)
; Нх
.р
. = -3267,7 кДж/моль (8.12) 2
Сложим (8.10) и (8.11) и вычтем из суммы (8.12):
6С
(графит)
+ 6О
2(г)
+ 3Н
2 (г) + 3/2О
2 (г) - C
6H
6(ж) -
15 О
2(г) =
6СО
2(г)
+ О
2(г) =
6СО
2(г)
+
2
3Н
2О
(ж) - 6CO
2(г) -3H
2O
(ж);
Нх
.р
. = {6∙(-393,51) + 3∙ (-285,84) – (-3267,7)} кДж/моль,
или:
6С
(графит) + 3Н
2(г) = C
6H
6(ж);
Нх
.р
. = {6∙(-393,51) + 3∙ (-285,84) – (-3267,7)} кДж/моль (8.13)
Проанализировав (8.13), видим, что энтальпия реакции
(в данном случае это энтальпия образования жидкого бензола) равна разности между суммой теплот сгорания исходных веществ и суммой теплот сгорания продуктов реакции, с учетом количества моль каждого вещества, или
:
H
обр
0 C
6H
6 = 6
H
сгор
С + 3
H
сгор
Н
2 -
H
сгор
C
6H
6
(8.14)
Подставляя в (8.14) значения теплот сгорания веществ, участвующих в реакции, получаем:
H
обр
0
C6
H6
= 6∙(– 393,51) + 3∙(– 285,84) – (-3267,7) = +49,12 кДж/моль;
Энтальпию не только этой, но и любой другой реакции можно определить как сумму теплот сгорания исходных веществ за вычетом суммы теплот сгорания продуктов реакции, с учѐтом количества моль каждого вещества
. Это следствие из закона Гесса применительно к теплоте образования.
Величины теплоты образования важны не только для энергетических расчетов, но и для соблюдения техники безопасности. В приведенном выше примере энтальпия образования бензола – величина положительная. Вещества, у которых энтальпия образования – положительная величина, неустойчивы, так как для того, чтобы их получить, энергию пришлось затратить. Вещества, у которых энтальпия образования – большая положительная величина, поскольку при разложении их на простые вещества теплота, затраченная на их образование, выделится обратно. Это, например, ацетилен (+227 кДж/моль), йодистый азот (+230 кДж/моль), нитроглицерин (+1430 кДж/моль), тротил (тол, или тринитротолуол) (+980 кДж/моль) и т.д.
8.5. Вопросы и задания
1. Откуда берется теплота при химических реакциях?
2. Что такое теплота стандартная энтальпия образования вещества?
3.  Сколько литров метана нужно сжечь при нормальных условиях, чтобы получить 500 кДж теплоты? Реакция сгорания метана: CH4
(г)+ 2O2
(г) = CO2
(г) + 2 H2
O; H
-890,31 кДж/моль. Сколько литров метана нужно сжечь при нормальных условиях, чтобы получить 500 кДж теплоты? Реакция сгорания метана: CH4
(г)+ 2O2
(г) = CO2
(г) + 2 H2
O; H
-890,31 кДж/моль.
4. Сколько теплоты выделяется при получении 2 моль этанола в термохимическом процессе:
С2
Н4
(г)+Н2
О(ж)→С2
Н5
ОН(ж); H
-44 Дж/моль?
5. Определите энтальпию реакции: 2 S + 3 O2
= 2 SO3,
если известны энтальпии реакций: а) S + O2
= SO2
; H
-297 кДж/моль; и б) SO2
+ 0,5 O2
= SO3
; H
-396 кДж/моль
6. Вычислите стандартную энтальпию образования (∆Н обр.) анилина C6
H7
N(ж)
, если известна его теплота сгорания (∆Н сгор.) при Т= 298К и Р=1,0133∙10-5
Па (-3396,2 кДж/моль). Продуктами сгорания являются CO2(г)
, H2
O(ж)
и N2г)
. Теплоты сгорания С и Н2
и N2
соответственно равны:
 С(граф) + О2
= СО2
(г); Hсгор
– 393,51 кДж/моль С(граф) + О2
= СО2
(г); Hсгор
– 393,51 кДж/моль
Н2
+ 1/2О2
= Н2
О(ж); Hсгор
– 285,84 кДж/моль
1/2N2
+ 1/2О2
= NО(г); Hсгор
+90,3 кДж/моль
7. Газообразный этиловый спирт С2
Н5
ОН(г) можно получить при взаимодействии этилена С2
Н4
(г) и водяных паров. Напишите термохимическое уравнение этой реакции и вычислите ее энтальпию. Стандартные энтальпии образования газообразного этилового спирта, этилена и воды равны соответственно –235,31; +52,28 и –241,83 кДж/моль.
8. При сгорании 9,3 фосфора выделяется 229,5 кДж теплоты. Определите энтальпию образования оксида фосфора Р2
О5
.
9. Чем тепловой эффект отличается от энтальпии?
10. Можно ли, исходя из энтальпии образования, определить, взрывоопасно ли вещество?
Лекция 9. Химическое и фазовое равновесие
9.1. Химическое равновесие
Все химические процессы можно разбить на две группы – это обратимые и необратимые реакции
. Необратимые реакции протекают до конца, то есть до полного израсходования исходных веществ, только в одном направлении[13]
. Обратимые реакции идут не до конца, так как они могут протекать и в прямом, и в обратном направлении.
Накопление продуктов реакции повышает возможность соударения их молекул между собой и разложение их на исходные вещества.
Рассмотрим, например, реакцию образования йодистого водорода.
H2
+I2
= 2HI;
Скорость прямой реакции по закону действия масс равна:
V1
= k1
[H2
][I2
];
Скорость обратной реакции тоже описывается законом действия масс:
V2
= k2
[HI]2
По истечении некоторого времени скорости прямой и обратной реакции сравняются.
V1
= V2
;
или k1
[H2
][I2
] = k2
[HI]2
Разделив обе части на k2
[H2
][I2
],
получаем
k
1
[HI
]2
K
p
(9.1)
 k
2 [H
2] [I
2] k
2 [H
2] [I
2]
9.2. Константа равновесия
Величину Kр
называют константой химического равновесия.
В правой части уравнения стоят те концентрации взаимодействующих веществ, которые устанавливаются при равновесии.
Дальнейшие непрекращающиеся переходы исходных веществ в продукты реакции, и, наоборот, к изменению концентраций всех компонентов смеси приводить не будут. Следовательно, при равновесии система реагирующих веществ характеризуется постоянством макроскопических свойств (например, неизменными равновесными концентрациями всех компонентов системы), а равновесие в системе является динамическим, подвижным
.
В случае гетерогенных реакций
в выражение для константы равновесия, так же, как и в законе действия масс, входят концентрации только тех веществ, которые находятся в газовой фазе или в растворе.
Например, для обратимой реакции:
СО2
(г) + С(тв) ↔ 2СО(г)
выражение для константы равновесия имеет вид: [CO
]2
 K
(9.2) K
(9.2)
[CO
2
]
Для реакций, протекающих в газовой фазе, при вычислении константы равновесия удобно пользоваться парциальными давлениями
реагирующих веществ. Например, для реакции: 2СО2(г)
↔2СО(г)
+О2(г)
Выражение для константы равновесия имеет вид:
P
CO
2 P
O
2
 K
р
P
CO
2
2 (9.3) K
р
P
CO
2
2 (9.3)
Где Р – парциальное давление каждого газообразного компонента смеси.
На практике абсолютно необратимых реакций не бывает, любая реакция всегда протекает как в прямом, так и в обратном направлении. Поэтому равновесие устанавливается и в «необратимых» реакциях, хотя в таких случаях оно очень сильно сдвинуто влево, в сторону исходных веществ.
Так, при реакциях, протекающих с образованием осадков, константа равновесия называется произведением растворимости.
Например, при образовании осадка сернокислого бария в результате реакции ионного обмена установится равновесие: BаSO
4 (ТВ) ↔ Ba
2+ + SO
42-;
Константой равновесия данного процесса, равновесие в котором сильно сдвинуто влево, является соотношение:
K
 (9.4) (9.4)
[BaSO
4
]
Знаменатель дроби – концентрация твердой соли – представляет собой постоянную величину, которую можно ввести в константу равновесия и обозначить ПР. Тогда получаем:
[Ba2+
]∙ [SO4
2-
] = ПР;
Равновесие наступает и при образовании малодиссоциированного соединения. Например, встречаясь в растворе, ион водорода и ацетатион образуют слабую уксусную кислоту. Равновесие в данном процессе сильно сдвинуто влево.
СН3
СООН ↔
СН3
СОО-
+ Н+
Константой равновесия данного процесса является соотношение:
[H
] [CH
3
COO
]
 K
д
(9.5) K
д
(9.5)
[CH
3
COOH
]
В данном случае константа равновесия называется константой диссоциации Кд; в квадратных скобках приведены равновесные молярные концентрации ионов водорода, ацетат-аниона и уксусной кислоты
В окислительно-восстановительных процессах константа равновесия зависит от окислительно-восстановительных потенциалов:
lgK
(9.6)
 где где  - потенциал окислителя, - потенциал окислителя, а n = число электронов, участвующих в процессе. - потенциал окислителя, - потенциал окислителя, а n = число электронов, участвующих в процессе.
Понятие «равновесие» применимо к любому химическому процессу. Более того, в любой изолированной химической системе рано или поздно наступает состояние равновесия. В практических целях часто бывает необходимо «сдвинуть» это равновесие в сторону желательной реакции. Это можно сделать, пользуясь
сформулированным французским химиком Анри де Ле Шателье
(18501936) в 1884-1885 гг. принципом подвижного равновесия
(немецкий физик К.Ф.Браун дал этому принципу теоретическое обоснование в 1887 году).
9.3. Принцип Ле Шателье
Если на систему, находящуюся в состоянии подвижного устойчивого равновесия, воздействовать извне, изменяя условия, определяющие положение этого равновесия, в ней начнутся процессы, направленные на ослабление производимого воздействия.
Другими словами, если из системы удалять один из компонентов, система будет отвечать на такое внешнее воздействие, «поворачиваясь» в направлении той реакции, при которой синтезируется удаляемое вещество. Если мы хотим, чтобы шла реакция, при которой выделяется тепло, мы должны охлаждать реакционную смесь. Равновесие в системе сдвинется в сторону той реакции, которая идет с понижением давления, если мы будем повышать давление в системе. Принцип Ле Шателье дает возможность управлять реакциями, которые имеют большое значение в промышленности (например, синтез аммиака, пиролиз и крекинг углеводородов, и многие другие процессы).
Процесс изменения концентраций реагирующих веществ, вызванный нарушением равновесия, называется смещением, или сдвигом равновесия
. Если при этом происходит увеличение концентраций продуктов реакции, говорят, что равновесие сдвигается вправо
, то есть в направлении прямой реакции; при увеличении концентраций исходных веществ равновесие сдвигается влево
, в сторону обратной реакции.
Могут иметь место следующие случаи:
1)
При увеличении концентрации какого-либо из веществ, участвующих в реакции, равновесие смещается в сторону расхода этого вещества. При уменьшении концентрации какого-либо вещества равновесие смещается в сторону его образования.
Пусть, например, водород, йодистый водород и пары йода находятся в равновесии друг с другом при постоянной температуре и давлении. Введем в систему дополнительно некоторое количество водорода. По закону действия масс увеличение концентрации водорода повлечет за собой увеличение скорости прямой реакции. В результате концентрации йода и водорода будут уменьшаться, а концентрация йодистого водорода возрастать, что приведет к увеличению скорости обратной реакции. Через некоторое время вновь наступит равновесие, но уже при других равновесных концентрациях.
2)
При увеличении давления путем сжатия системы равновесие смещается в сторону уменьшения числа молекул (молей) газов, то есть в сторону понижения давления; при уменьшении давления равновесие сдвигается в сторону реакции, при которой число молекул газа возрастает, то есть в сторону увеличения давления.
Изменение давления нарушает равновесие только в реакциях, идущих в газовой фазе.
3)
При повышении температуры равновесие смещается в сторону эндотермической, а при понижении температуры – в сторону экзотермической реакции.
Знание принципов химического равновесия способствует прогрессу химической промышленности. Так, например, реакция получения аммиака из азота и водорода идѐт по уравнению:
N2(г)
+ 3H2(г)
↔ 2NH3(г)
; H
-46,2 кДж/моль
Применяя принцип Ле Шателье, можно предсказать оптимальные условия синтеза аммиака. Для того, чтобы сместить равновесие вправо, в сторону образования продуктов реакции, необходимо повышать концентрацию исходных веществ и выводить продукты из сферы реакции. Поскольку энтальпия прямой реакции меньше нуля, прямой процесс идѐт с выделением тепла, а обратный – с поглощением теплоты. Следовательно, чтобы сместить равновесие вправо, необходимо понижать температуру реагирующей смеси. Поскольку реакция происходит в газовой фазе, фактор давления тоже влияет на равновесие. Так как в прямом процессе число молей уменьшается (с 4 до 2), а в обратном увеличивается, для смещения равновесия вправо давление необходимо повышать. На практике синтез аммиака ведут при умеренной температуре 500о
С с применением катализатора, чтобы повысить скорость реакции (так как в данном случае для выгодного состояния равновесия требуется низкая температура). Процесс ведут при давлении 350 атмосфер, так как применение очень высокого давления требует специального и дорогого оборудования. В таких условиях только 30% исходных веществ превращается в аммиак. Аммиак отделяют от них методом сжижения, а не прореагировавшие водород и азот возвращают в сферу реакции с тем, чтобы максимально повысить выход продукции.
Принцип Ле Шателье распространяется не только на химические, но и на различные физико-химические равновесия, такие как кипение, кристаллизация или растворение
, а также на процессы, происходящие в живой природе.
9.4. Фазовое равновесие
Рассмотрим основные понятия фазового равновесия
– раздела физической химии, который рассматривает приложение законов термодинамики к изучению свойств гетерогенных систем.
В зависимости от внешних условий вещество может находиться в различных фазах, соответствующих его агрегатному состоянию. Изменение агрегатного состояния вещества называется фазовым переходом
. Это, например, испарение, конденсация, плавление, кристаллизация. Как и любой термодинамический процесс, фазовый переход протекает до установления в системе некоторого равновесного состояния, которое характеризуется постоянством температуры, давления и термодинамического потенциала (∆G = 0).
Фаза
– это совокупность материальных частиц системы, обладающих одинаковыми термодинамическими свойствами. Гомогенная система состоит из одной фазы, а гетерогенная представляет собой две или более фазы, отделенными друг от друга поверхностями раздела.
Число фаз системы может изменяться от единицы до бесконечности. Понятие фазы отличается от понятия агрегатного состояния своей конкретностью. Например, смесь воды, бензола, песка, медных опилок и паров воды бензола и воды над этой смесью представляет собой пятифазную систему
, состоящую из двух кристаллических, двух жидких и одной газообразной фаз. Совокупность песчинок – одна фаза (к), медные опилки – другая (к), вода – третья (ж), бензол – четвертая (ж), а смесь паров воды и бензола – пятая (г).
Другое важное понятие при рассмотрении фазового равновесия – компонент
. Компонент – это однородная по химическим свойствам часть термодинамической системы, которая может быть выделена из нее и может существовать изолированно неограниченное время. Так, например, однофазная система «раствор поваренной соли» является двухкомпонентной, хотя состоит фактически из молекул хлористого натрия и воды, а также катионов натрия и хлорид-анионов. Однако, из системы можно выделить в свободном состоянии только два компонента – молекулы поваренной соли и воды, а ионы натрия и хлора в свободном состоянии существовать не могут.
Между компонентами равновесной системы существует зависимость в виде взаимосвязи их концентраций. По этой причине вводится понятие числа независимых компонентов системы
, под которыми подразумевают минимальное число компонентов, достаточное для построения любой фазы системы. Число независимых компонентов системы Кн
равно общему числу ее компонентов К
за вычетом числа химических реакций, которые могут протекать в системе при данных условиях, и числа дополнительных условий, связывающих концентрации компонентов системы между собой.
Кн
= К - ∑С(сi
),
где последнее выражение отражает общее число функциональных зависимостей компонентов системы между собой.
Например, в системе, состоящей из произвольно взятых количеств хлорида аммония, аммиака и хлористого водорода, число независимых компонентов равно двум, так как равновесный состав ее в этом случае контролируется одной функциональной зависимостью – законом
действия масс, и
Кн
= 3 -1=2
.
А если на эту систему наложить дополнительное условие, например, равенства концентраций аммиака и хлористого водорода, то число независимых компонентов станет равным единице:
Кн
= 3 –2 =1
.
Третье важное понятие при рассмотрении фазового равновесия – это степень свободы
, под которой понимают возможность произвольного изменения (в определенных пределах) какого-нибудь параметра состояния (температуры, давления, концентрации компонентов и пр.) без нарушения фазового равновесия, то есть без изменения числа и вида фаз, находящихся в равновесии. Например, температура и давление сухого воздуха могут изменяться в широких пределах без изменения его газообразного состояния, то есть термодинамическая система «сухой воздух» имеет две степени свободы. А вот температура кипящей жидкости (двухфазная система, состоящая из жидкости и ее паров) может изменяться лишь при изменении давления. Если же повысить температуру жидкости, сохранив ее давление постоянным, то фазовое равновесие нарушится. Одна фаза системы – жидкая – прекратит свое существование. В последнем примере число степеней свободы системы равно единице (это или давление, или температура).
Между числом фаз, независимых компонентов и степеней свободы равновесной термодинамической системы существует зависимость, называемая правилом фаз
.
9.5. Правило фаз
Для любой равновесной термодинамической системы сумма числа степеней свободы С и числа фаз Ф равна сумме независимых компонентов Кн
и числа внешних факторов n, влияющих на физическое состояние системы. Или, математически,  (9.7) (9.7)
Значение n зависит от условий, в которых находится система. Если силовые поля (гравитационное, магнитное, электрическое), в которых находится система, постоянны, а изменяются лишь температура и давление, то для неконденсированных систем, содержащих газовую фазу (зависящих от значения давления) n = 2. Для неконденсированных систем, не содержащих газовой фазы и не зависящих вследствие этого от давления n = 1. Например, для сплавов правило фаз имеет вид: С + Ф = Кн + 1,
или С = Кн
+1 – Ф
.
При условии С =0 система называется безвариантной
. Это значит, что нельзя изменить ни один из параметров системы. При С = 1 (моновариантная
система) можно изменять один параметр состояния, а все остальные параметры зависят от первого. При С = 2 (бивариантная
система) можно изменять независимо друг от друга два параметра состояния, а все остальные параметры будут зависеть от этих двух.
Таким образом, правило фаз позволяет определить условия, при которых возможно сохранение данного фазового равновесия.
Основной инструмент изучения фазового равновесия в термодинамических системах – это так называемая фазовая диаграмма, или диаграмма состояния
. Фазовая диаграмма позволяет судить об агрегатных состояниях компонентов и их изменениях. Особенное значение имеют фазовые диаграммы при изучении свойств различных сплавов.
9.6. Диаграммы состояния
Диаграммы состояния получают экспериментально. Обычно для этого строят кривые охлаждения сплава с различным содержанием компонентов и по остановкам и точкам перегиба, вызванными различными тепловыми эффектами, определяют температуры, при которых происходят эти превращения. Для получения кривых охлаждения из двух металлов изучаемого сплава приготовляют ряд смесей различного состава. Каждую смесь расплавляют. Полученные жидкие сплавы медленно охлаждают, отмечая через определенные промежутки времени температуру остывающего сплава. По данным наблюдений строят графики – кривые охлаждения.
На диаграммах состояния по вертикальной оси откладывают температуру, а по горизонтальной – состав сплава (содержание одного из компонентов). Для сплавов из двух компонентов Х и У состав сплава относительно одного из компонентов обозначают точкой на соответствующем отрезке прямой, принятой за 100%.
Четырем упомянутым выше типам сплавов, а именно, механической смеси, твердому раствору с неограниченной и ограниченной растворимостью металлов друг в друге и химическому соединению, отвечают фазовые диаграммы соответствующего вида.
Рис. 9.1. Диаграмма плавкости механической смеси
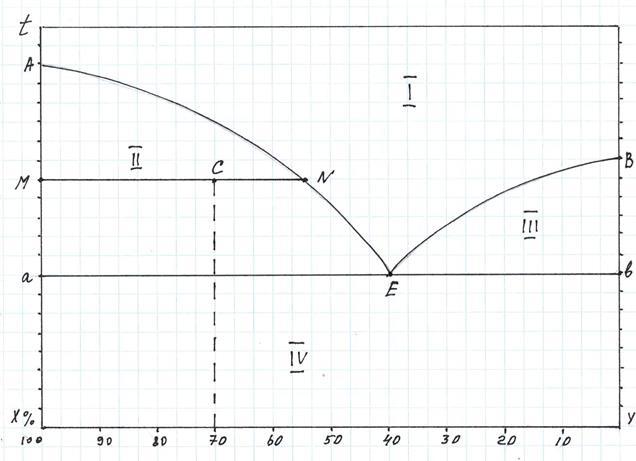
На рисунке 9.1 показана диаграмма состояния сплава, образующего механическую смесь индивидуальных компонентов. Точки А И В – это температуры плавления компонентов системы Х и У соответственно. В сплавах добавление одного компонента к другому понижает температуру его кристаллизации (затвердевания). Кривая АЕ показывает температуру кристаллизации металла Х из расплавов, богатых металлом Х, а кривая ВЕ – температуру кристаллизации металла У из расплавов, богатых металлом У. Число степеней свободы системы в момент ее равновесной кристаллизации в рассматриваемых случаях равно 1: С = 2 + 1 –2 = 1. Это значит, что произвольно можно изменять только один параметр состояния – температуру или состав сплава. Другой параметр устанавливается в зависимости от указанной на диаграмме состояния закономерности.
Точка Е принадлежит обоим кривым: из расплава, состав которого отвечает этой точке, кристаллизуются одновременно оба металла. Эта совместная кристаллизация происходит при самой низкой температуре. Отвечающий точке Е состав сплава называется эвтектическим составом
, или просто эвтектикой
(от греческого «эвтектикос» – хорошо плавящийся). Число степеней свободы в точке кристаллизации эвтектики равно нулю: С = 2 + 1 – 3. Это значит, что трехфазное равновесие в конденсированной двухкомпонентной системе возможно только при единственном значении состава и температуры.
Линии АЕ и ВЕ – это зависимости температуры начала кристаллизации двухкомпонентной системы от ее состава. Их называют линиями ликвидуса, или ликвидусом
(от латинского слова ―жидкий‖). Прямая аЕb представляет собой температурную зависимость конца кристаллизации двухкомпонентной системы. Ее называют линией солидуса, или солидусом
(от латинского слова «твердый»).
Область I, расположенная выше ликвидуса – это область жидкого состояния системы с числом степеней свободы, равным 2: С = 2+ 1 - 1. Это значит, что в таком состоянии в системе можно менять оба параметра состояния (температуру и состав).
Область II, расположенная между левой ветвью ликвидуса и солидусом, изображает двухфазное равновесное состояние системы, являющейся смесью расплава, насыщенного компонентом Х, и кристаллов металла Х.
Область III, расположенная между правой ветвью ликвидуса и солидусом, изображает двухфазное равновесное состояние системы, представляющей собой смесь расплава, насыщенного компонентом У, и кристаллов компонента У.
Область IV изображает кристаллическое состояние системы. В таком состоянии сплав представляет собой тонкую механическую смесь кристаллов металлов Х и У. Число степеней свободы системы в таком состоянии равно 1: С = 2 + 1 –2 = 1. Этой степенью свободы является температура.
При термодинамически обратимом охлаждении расплава на всем пути изменения его температуры от начала кристаллизации и до образования эвтектики между его твердой и жидкой фазами соблюдается термодинамическое равновесие. Линию, соединяющую на диаграмме точки, соответствующие состояниям равновесных фаз, называют нодой
. Это, например, линия MN, соединяющая точку М, соответствующую состоянию твердой фазы (кристаллы металла Х) с точкой N, соответствующей состоянию расплава состава l, равновесного при данной температуре tc
с указанной твердой фазой. Ноды используют для предварительного определения массы кристаллов, образующихся при охлаждении расплавов. Расчет ведут по правилу рычага:
Отношение массы твердой фазы Мкр
(кристаллов) по отношению к массе жидкой фазы Мр
(расплава) равно отношению отрезка ноды, соответствующего изменению состава расплава при его охлаждении, к отрезку ноды, соответствующему исходному состоянию расплава.
Другими словами, Мкр
:Мр
= СN:МС.
Отсюда следует, что отношение массы образовавшихся кристаллов Мкр
к исходной массе расплава Мисх
равно отношению отрезка ноды, соответствующего изменению состава расплава при его охлаждении, ко всей ноде, или Мкр
:Мисх
= СN:MN.
Пример 1.
Найти число степеней свободы системы свинец-висмут, если из расплава Pb – Bi будут выпадать кристаллы висмута.
Под числом степеней свободы в равновесной гетерогенной системе понимают условия (температуру, давление, концентрацию компонентов), которые можно произвольно изменять, не нарушая равновесия системы и не изменяя число фаз в системе. Число степеней свободы системы рассчитывается с помощью правила фаз:
С = Кн – Ф + 1
(для систем, не содержащих газовой фазы).
В данной системе 2 независимых компонента (свинец и висмут) и две фазы – расплав и кристаллы висмута. Число степеней свободы в этой системе равно С = 2 – 2 + 1 = 1. Эта система имеет одну степень свободы, и можно произвольно изменять либо температуру, либо концентрацию компонентов системы, не нарушая ее равновесия.
Пример 2.
Сплав содержит 24% кадмия и 76% висмута. В 1000 г сплава содержится 400 г висмута в виде кристаллов, вкрапленных в эвтектику.
Рассчитайте состав эвтектики.
Эвтектика – неоднородная механическая смесь, состоящая из мелких кристаллов двух компонентов сплава, образовавшаяся при одновременной кристаллизации обоих компонентов. Масса эвтектики равна 600 г (1000 – 400). Масса содержащегося в сплаве кадмия равна 240 г, а висмута – 760 г. Следовательно, эвтектика содержит 240 г кадмия и 760 – 400 = 360 г висмута. Процентное содержание кадмия в эвтектике равно 240:600 (х100%) = 40%; процентное содержание висмута в эвтектике равно 360:600 (х100%) = 60%.
Пример 3.
Найдите состав твердой и жидкой фаз в системе, содержащей 60% свинца и 40% олова при 2000
С. Какова масса твердой фазы, выделившейся при этой температуре из 5 кг сплава? Диаграмма плавкости системы приводится ниже.
Рис. 9.2. Диаграмма плавкости «свинец-олово».

На диаграмме плавкости сплава свинца с оловом данному составу и данной температуре соответствует точка К, через которую проводим прямую, параллельную оси абсцисс, до пересечения с кривой АЕ и ординатой, соответствующей чистому свинцу. Точка F отвечает составу жидкой фазы, а точка N – составу твердой фазы. Жидкая фаза имеет состав 58% олова и 42% свинца. Твердая фаза представляет собой чистый свинец.
Масса твердой фазы Мт и жидкой фазы Мж в сплаве данного состава и при данной температуре определяют по правилу рычага: массы твердой и жидкой фаз обратно пропорциональны длинам отрезков между точкой, выражающей данное состояние системы, и точками, определяющими состав твердой и жидкой фаз.
Мт: Мж = KF:NK. Общая масса сплава 5 кг, тогда Мж = 5 – Мт. NK = 40, KF = 15. Тогда Мт:5-Мт = 15: 40; 40Мт = 75 –15Мт; 55Мт = 75; Мт = 75:55 = 1,36 кг. Таким образом, из 5 кг сплава при температуре 200о
С выделяется 1, 36 кг свинца (твердой фазы).
Твердые растворы
образуют компоненты сплавов, неограниченно растворяющиеся друг в друге как в жидком, так и в твердом состоянии и не образующие при этом химических соединений. Например, серебро и золото образуют при сплавлении твердый раствор. Сплаву Ag – Au отвечает следующая диаграмма плавкости (рис. 10).
Кривая АВС отвечает температуре плавления сплавов; кривая ADC – температуре затвердевания сплавов. Вид кривых плавления (верхней и нижней) обусловлен тем, что кристаллы, выделяющиеся при охлаждении расплава, всегда содержат оба компонента (кроме кристаллизации чистых серебра и золота).
Области I на диаграмме состояния отвечает расплав, области II – сосуществование расплава и кристаллов твердого раствора, области III – твердый раствор.
Пример 4:
Определить температуру затвердевания и плавления сплава, содержащего 25% серебра и 75% золота.
Сплаву, содержащему 25% серебра и 75 % золота, соответствует точка К на оси абсцисс. Из точки К проводим прямую, параллельную оси ординат, до пересечения с кривой плавления и затвердевания в точках В и D. Из этих точек проводим прямые, параллельные оси абсцисс, до пересечения с осью ординат в точках B1
и D1
. Точка D1
соответствует температуре затвердевания сплава, которая равна 1025о
С. Температура полного плавления сплава равна 1045о
С (точка В1
).
Рис. 9.3. Диаграмма плавкости твердого раствора (золото-серебро)
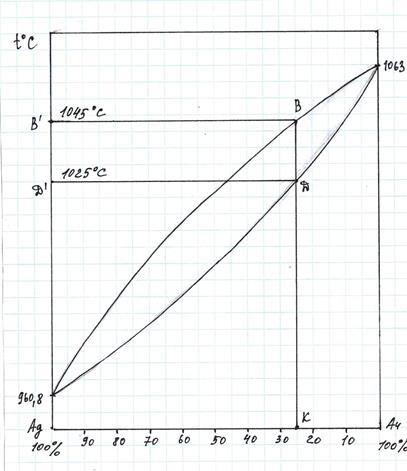
Диаграмма состояния для сплавов с ограниченной растворимостью компонентов друг в друге имеет более сложный вид и здесь не рассматривается.
Интерметаллические соединения
образуются в том случае, когда компоненты сплава химически взаимодействуют между собой и в жидком состоянии полностью растворимы друг в друге, а в твердом состоянии совершенно нерастворимы. О рассмотренных ранее диаграмм эта диаграмма отличается наличием максимума на кривой ликвидуса. Диаграммы плавкости этого типа представляют собой как бы сочетание двух эвтектических диаграмм первого типа.
Рис. 9.4. Диаграмма плавкости интерметаллического соединения (свинецмагний)

На рисунке 11 приведена диаграмма состояния системы магний – свинец. Эта система образует интерметаллическое соединение состава
Mg2
Pb. Абсцисса точки максимума указывает состав интерметаллического соединения и отвечает температуре его плавления. На диаграмме имеются две эвтектики Е1
и Е2
. Эвтектика Е1
представляет собой смесь кристаллов магния и Mg2
Pb, а эвтектика Е2 – кристаллов свинца и Mg2
Pb.
Если компоненты системы способны образовывать несколько химических соединений, то диаграмма будет как бы составлена из трех, четырех и более отдельных диаграмм первого типа.
Области I на диаграмме отвечает жидкий сплав, областям II –V – равновесия жидкого сплава и соответствующих кристаллов (в области II – кристаллы магния, в областях III И IV – кристаллы Mg2
Pb, в области V – кристаллы свинца). Областям VI – IX соответствуют твердые сплавы (магний + эвтектика Е1
в области VI, Mg2
Pb + эвтектика Е1
в области VII, Mg2
Pb + эвтектика Е2
в области VIII, свинец и эвтектика Е2
в области IX). Пример 5.
По диаграмме плавкости системы Mg – Pb определите формулу интерметаллического соединения, образуемого этими металлами. Какова масса химического соединения, содержащегося в 500 г сплава состава 40% свинца и 60% магния?
Максимум на кривой ABCDM (точка С) отвечает температуре плавления интерметаллического соединения, образованного магнием и свинцом. Температура плавления интерметаллического соединения равна 551о
С. Из диаграммы плавкости видно, что интерметаллическое соединение содержит 80% свинца и 20% магния. Обозначим формулу интерметаллического соединения через Mgх
Pbу
. Атомная масса магния равна 24,305, а атомная масса свинца – 207,29. Тогда х:у = 20/24,305:80/207,29 = 0,82:0,39 = 2:1. Формула соединения - Mg2
Pb.
В сплаве магния больше, чем в составе химического соединения. Следовательно, свинец полностью входит в состав соединения. В 500 г сплава содержится 500∙0,4 = 200 г свинца. По массе свинца определяем массу химического соединения: 200:0,8 = 250 г.
Мы рассмотрели простейшие, но наиболее важные диаграммы состояния. Для многих сплавов и других многокомпонентных систем диаграммы состояния носят значительно более сложный характер. Ряд металлов и сплавов испытывают превращения в твердом состоянии, переходя из одной модификации в другую (например, α, β, γ и δ- железо).
Для технически важных систем диаграммы состояния хорошо изучены и приводятся в специальной литературе. Они служат научной основой при подборе сплавов, обладающих заданными свойствами, при разработке новых сплавов и методов их термической обработки. Примером системы, имеющей огромное значение, служит система железо-углерод. В 1868 году русский металлург Д.К Чернов впервые указал на зависимость микроструктуры стали от определенных температур («критических точек»). Изучение диаграмм состояния углерод-железо положило начало материаловедению
– науки о строении и свойствах металлов и сплавов.
В дополнение к термическому анализу часто проводят микроскопическое металлографическое
исследование отшлифованных и отполированных образцов различных металлов и сплавов, обработанных травителем. Травитель выбирают таким образом, чтобы он растворял преимущественно один компонент сплава. Изучение структуры сплава, выявленной таким образом, помогает определить тип взаимодействия металлов, входящих в состав сплава.
9.7. Вопросы и задания:
1.
При синтезе аммиака по схеме: N2
+3H2
↔ 2NH3
равновесие установилось при следующих концентрациях реагирующих веществ: [N2
] =2,5 моль/л; [H2
]= 1,8 моль/л и [NH3
] = 3,6 моль/л. Рассчитайте константу равновесия.
2.
Объѐмный состав реакционной смеси: 2СО2(г)
↔ 2СО(г)
+ О2(г)
в момент равновесия составлял: 88,72% СО2; 7,52% СО и 3,76%О2. Определите константу равновесия этой реакции, если общее давление в системе равно 1,0133∙105
Па.
3.
Как следует изменить температуру, давление и концентрацию реагентов для смещения равновесия в системе: СаСО3(к)
↔СаО(к)
+СО2(к)
; H
>0 в сторону продуктов реакции?
4.
 В какую сторону сместится равновесие в системе: 2NO(г)
+Cl2(г)
↔2NOCl(г)
; H
73,6 кДж
при понижении давления и одновременном повышении моль В какую сторону сместится равновесие в системе: 2NO(г)
+Cl2(г)
↔2NOCl(г)
; H
73,6 кДж
при понижении давления и одновременном повышении моль
температуры?
5.
Как называется неоднородная механическая смесь, состоящая из мелких кристаллов двух компонентов сплава, образовавшаяся в результате одновременной кристаллизации обоих компонентов при самой низкой температуре?
6.
Какую систему образуют компоненты, неограниченно растворимые друг в друге в жидком состоянии и химически взаимодействующие между собой?
7.
Что образуется, когда компоненты системы неограниченно растворяются друг в друге в жидком состоянии и полностью растворимы в твердом состоянии?
8.
Как называется кривая температуры плавления на фазовой диаграмме двухкомпонентной системы?
9.
Определите число степеней свободы в гетерогенной системе свинецолово, если из расплава выпадают кристаллы олова.
10.
Из скольких фаз состоит двухкомпонентная система, не содержащая газовой фазы, если число ее степеней свободы равно единице?
Лекция 10. Скорость реакции и методы ее регулирования
Скорость химической реакции зависит от многих факторов. Главными из них являются природа реагирующих веществ
, концентрация реагирующих веществ
, температура реакционной среды
, энергия активации и катализатор
.
10.1 Природа реагирующих веществ
Две реакции одного типа, - окисление кислородом воздуха оксида азота NO:
2NO + O2
→2NO2
(10.1)
и метана CH4
:
СН4
+ О2
→ СО2
+ Н2
О (10.2)
Протекают в нормальных условиях с разными скоростями. Скорость реакции (10.1) достаточно велика, тогда как метан в (10.2) при комнатной температуре практически не окисляется. Различие в скоростях однотипных реакций в одинаковых условиях зависит исключительно от природы реагирующих веществ. Объясняется это различной способностью разных реагирующих частиц к активным соударениям. Влияние природы реагирующих веществ на скорость реакции пока ещѐ плохо изучена; количественной характеристикой этого влияния является константа скорости реакции. Константы скорости различных реакций определяют экспериментально. Размерность константы скорости реакции первого порядка обратна размерности времени, то есть сек-1
. Константа равновесия реакции второго порядка имеет размерность л∙моль-1
∙сек-1
, а константа равновесия реакции третьего порядка – л2
∙моль-2
∙сек-1
10.2. Концентрация реагирующих веществ
Зависимость скорости химической реакции от концентрации реагирующих веществ определяется законом действующих масс, или кинетическим уравнением (7.2).
Например, в химической системе,
где в газовой фазе протекает следующая реакция: 2СО(г) + О2
(г) → 2СО2
(г). Нас интересует, как изменится скорость прямой реакции образования диоксида углерода, если общее давление в системе увеличить в два раза. Рассуждаем следующим образом: при повышении давления в 2 раза объѐм реагирующей смеси уменьшается в 2 раза (так как PV=const), значит, концентрация каждого из реагирующих веществ увеличится в 2 раза.
Кинетическое уравнение для данной прямой реакции имеет вид:
V=k[CO]2
∙[О2] (10.3)
Если обозначить начальные концентрации моноксида углерода СО и кислорода О2
как a и b соответственно, то отношение конечной скорости реакции v2
(после повышения давления) к начальной скорости реакции (до повышения давления) v1
будет иметь вид:
V
2
k
(2a
)2
2b
2
 2
2 2 8 2
2 2 8
V
1
ka b
Следовательно, при повышении давления в сфере реакции в два раза скорость реакции возрастѐт в 8 раз.
10.3. Температура реакционной среды
При повышении температуры скорость реакции резко увеличивается, так как возрастает скорость хаотического движения реагирующих частиц и, как следствие, количество соударений. Голландский ученый Якоб Вант Гофф показал, что при повышении температуры на 10 градусов скорость большинства реакций увеличивается в 2 – 4 раза в соответствии со следующей полуэмпирической зависимостью:
 ,
(10.4) ,
(10.4)
1
где v – скорость реакции; t – температура в градусах Цельсия;  - температурный коэффициент (2-4). - температурный коэффициент (2-4).
Так, например, если температура реакционной среды повышается с 20
о о
С до 60 С, а температурный коэффициент реакции равен 3, то скорость данной реакции возрастѐт в соответствии с зависимостью Вант Гоффа
(10.4):  81,
в 81 раз. 81,
в 81 раз.
1
Между скоростью протекания химической реакции V и временем еѐ протекания t существует следующая зависимость:
 1 1
V
t
(10.5)
Если реакция протекает при температуре Т1
и Т2
, то эта зависимость принимает вид:
V t

V
T
1 t
T
2 (10.6)
При этом правило Вант-Гоффа можно записать в виде: t
1

t
2
(10.7)
При повышении температуры возрастает вероятность соударений, а это значит, что константа скорости реакции зависит от температуры так же, как и скорость процесса. По правилу Вант-Гоффа, эта зависимость
 k
2
имеет вид: .
Более точную зависимость константы скорости от k
2
имеет вид: .
Более точную зависимость константы скорости от
k
1
температуры установил шведский ученый Сванте Аррениус в 1889 году:
  k Ae
RTEa
,
или lnk
Ea
ln A
(10.8) RT k Ae
RTEa
,
или lnk
Ea
ln A
(10.8) RT
где А – предэкспоненциальный множитель, Еа
– энергия активации химической реакции, R – универсальная газовая постоянная, равная 8,3144103
Дж/кмольК; Т – температура в градусах Кельвина, е – основание натурального логарифма .
10.4. Энергия активации
Не все молекулы при данной температуре обладают одинаковой энергией и движутся с одинаковой скоростью. Существует распределение молекул по скоростям, а значит и по энергии. Только некоторая часть молекул движется с очень малой или очень большой скоростью, но большинство молекул движется с некоторой средней скоростью (рис. 10.1). Реагировать может только та часть молекул, в которых запасенная энергия выше некоторого определенного предела для каждой конкретной реакции (эта область на Рис.10.1 заштрихована).
Рис. 10.1. Распределение молекул по энергии при определенной температуре.

По горизонтальной оси отложена энергия молекул. На вертикальной оси показано количество молекул данной энергии. Значение Emin
представляет собой некоторую минимальную энергию, которой должна обладать молекула, чтобы вступить в некую химическую реакцию. Количество таких «энергичных» молекул примерно пропорционально заштрихованной площади под кривой. Пусть в химической системе протекает условная реакция A+B=C. В тот момент, когда «энергичная» молекула А вступает в химическое взаимодействие с молекулой B старые химические связи уже почти разрушились, а новые, характерные для молекулы C, еще не успели вполне сформироваться. В такой момент вещество, заключенное в этих молекулах, неустойчиво и имеет высокую энергию. Подобное состояние в химической реакции называется переходным состоянием, или активированным комплексом
(когда молекула представляет собой нечто «среднее» между молекулами А, B и C). Энергией активации
Еа
называют ту энергию, которая необходима для превращения реагирующих веществ в активированный комплекс. Не следует путать значения Еа и Еmin
из рис. 10.1. Во-первых, по достижении молекулами энергии Еmin
скорость реакции еще настолько мала, что такую реакцию мы вряд ли могли бы наблюдать. Во-вторых, энергия активации Еа - это разница между средней
энергией исходных веществ и средней
энергией того же вещества, уже находящегося в переходном состоянии (при этом переходные состояния тоже рассматриваются как молекулы активированного комплекса, пусть даже и необычные). В отличие от Emin
, которую можно представить для отдельной молекулы, энергия активации Еа может быть определена только для ансамбля реагирующих молекул. Термин «энергия активации» для отдельной молекулы не имеет никакого смысла. Энергия активации не зависит от температуры, однако при повышении температуры становится всѐ больше «энергичных» молекул, которые способны преодолеть активационный барьер
, разделяющий начальное и конечное состояние вещества в химической реакции.
Энергия активации прямой и обратной реакции связаны меж собой соотношением:
E
a
(пр
)  (пр
) (10.9) (пр
) (10.9)
Где Еа(пр)
– энергия активации прямой реакции; Еа(обр)
– энергия активации обратной реакции; H
(пр
)- энтальпия прямой реакции.
Активационный барьер
у химических реакций определяет само существование нашего мира. Если бы реакция окисления кислородом воздуха белков, углеводов, металлов шла безактивационно, все леса, продукты сельского хозяйства, металлы, да и мы сами давно превратились бы в ржавчину, углекислоту и воду. Энергия активации делает эти реакции при обычной температуре такими медленными, что мы их даже не замечаем. При повышении температуры окислительная способность кислорода резко возрастает. Так, например, небольшие количества кислорода, растворенного в котловой воде, способны привести к быстрой коррозии и выходу из строя оборудования тепловых электростанций, потому что при температуре перегретого пара всѐ больше и больше молекул кислорода преодолевают активационный барьер.
10.5. Катализаторы
Катализаторами называются вещества, ускоряющие химическую реакцию (см. Лекцию 5 «Катализаторы и каталитические системы»). Любая реакция характеризируется определѐнным активационным барьером, который должны преодолеть молекулы исходных веществ для перехода в продукты реакции, а также соответствующим механизмом реакции. При вводе в систему катализатора реакция протекает по другому механизму, с более низким активационным барьером. Следовательно, этот барьер может преодолеть большее число реагирующих частиц, что приводит к ускорению протекания реакции.
10.6. Вопросы и задания
1. Какие факторы влияют на скорость химической реакции?
2. Что такое энергия активации?
3. Сформулируйте полуэмпирическое правило Вант-Гоффа о зависимости скорости химической реакции от температуры.
4. Во сколько раз возрастѐт скорость химической реакции: 4NH3
+5O2
↔ 4NO+6H2
O, если увеличить концентрации исходных веществ в два раза?
5. Как изменится скорость химической реакции: СО(г)
+Cl2(г)
↔COCl2(г)
при увеличении давления в системе в 5 раз?
6. Во сколько раз быстрее пойдет химическая реакция, если еѐ температурный коэффициент равен 3, а температура в системе повысилась с 20о
С до 50о
С?
7. Как изменится скорость реакции при охлаждении системы от 100о
С до 80о
С, если температурный коэффициент равен 2?
8. При 80о
С реакция заканчивается за 20 секунд. Сколько времени будет идти эта реакция при 20о
С, если температурный коэффициент равен 2?
9. Константы скорости реакции при 273К и 298К равны соответственно 1,17 л∙моль-1
∙мин-1
. Найдите температурный коэффициент этой реакции.
10. Рассчитайте энергию активации химической реакции при 273К, если константа скорости реакции равна 4,04∙10-5
, а предэкспоненциальный множитель А равен 7,012∙106
сек-1
.
Лекция 11. Колебательные реакции
Особым типом химических процессов являются колебательные реакции
. Они протекают в довольно сложных реакционных системах, и для таких реакций очень важным оказывается термодинамический фактор. В таких системах возможно протекание ряда последовательных реакций, которые характеризуются различной скоростью. Взаимное наложение нескольких таких реакций, продукты которых могут оказывать каталитическое либо ингибирующее воздействие, проводит к тому, что в реакционной среде поочередно накапливается то один, то другой компонент. В случае интенсивно окрашенных веществ в значительных концентрациях колебательные реакции легко наблюдать.
11.1. Колебательные процессы в химии
Впервые, по имеющимся ныне данным, колебательную химическую реакцию, проявляющуюся в виде периодических вспышек свечения при окислении паров фосфора, наблюдал Роберт Бойль в конце XVII века. Эти вспышки потом неоднократно описывали другие исследователи. Химик Розеншельд в 1834 г. заметил, что небольшая колба, содержащая немного фосфора, в темноте испускает довольно интенсивный свет, причем это свечение регулярно повторяется каждую седьмую секунду. М. Жубер в 1874 г. наблюдал периодическое образование «светящихся облаков» в пробирке с парами белого фосфора. Позднее А. Центнершвер исследовал влияние давления воздуха на периодические вспышки фосфора: он установил, что частота вспышек начинается с 20 секунд и растет с понижением давления. В то же время Т. Торп и А. Таттон наблюдали в запаянном стеклянном сосуде периодические вспышки в процессе окисления триоксида фосфора. В 1896 г. немецкий химик Р. Лизеганг, экспериментируя с химикатами для фотографии, обнаружил, что если капнуть раствором нитрата серебра на стеклянную пластину, покрытую желатиной, содержащей дихромат калия, то продукт реакции, выпадая в осадок, располагается на пластинке концентрическими окружностями. Лизеганг увлекся этим явлением и почти полвека занимался его исследованием. Нашлось и практическое его применение: «кольца Лизеганга» использовали для украшения различных изделий с имитацией яшмы, малахита, агата и т.п.
В дальнейшем были открыты гетерогенные колебательные реакции на границе раздела двух фаз. Из них наиболее известны реакции на границе металл–раствор, получившие специфические названия – «железный нерв» и «ртутное сердце». Первая реакция растворения железной проволоки в азотной кислоте получила свое название из-за внешнего сходства с динамикой возбужденного нерва, замеченного В.Ф. Оствальдом. Вторая реакция разложения пероксида водорода Н2
О2
происходит на поверхности металлической ртути. При этом на поверхности ртути периодически образуется и растворяется пленка оксида. Колебания поверхностного натяжения ртути обусловливают ритмические пульсации ртутной капли, напоминающие биение сердца.
Природа колебаний была совершенно непонятна, поскольку химическая термодинамика и кинетика как науки еще не существовали, и никто не имел представления о том, какие законы управляют химическими реакциями. Лишь во второй половине XIX века, когда законы термодинамики и кинетики были открыты, интерес к колебательным реакциям и методам их анализа вспыхнул вновь. Причиной колебаний стали считать неравновесные фазовые переходы в гетерогенных системах, то есть такие колебания объяснялись примерно так: реакция идет, затем тормозится своими продуктами, которые удаляются в другую фазу, и реакция идет снова. В гомогенных же системах никаких макроскопических перемещений не наблюдается, поэтому возникновение колебаний в гомогенных системах долгое время считалось невозможным.
Возможность концентрационных колебаний в чисто гомогенных системах была сначала предсказана теоретически. В 1910 году итальянец А. Лотка предложил математическую модель, из которой следовала такая возможность. В основу этой модели был положен закон действующих масс. Споры вокруг модели Лотка не прекратились и тогда, когда в 1921 году англичанин Уильям Брей опубликовал статью, в которой достаточно подробно описал суть первой колебательной жидкофазной реакции разложения пероксида водорода, катализируемого йодатом. Реакция, которую впоследствии стали называть реакцией Брея-Либавского
, протекает следующим образом: 1) 5H2
O2
+ I2
→ 2HIO3
+ 4H2
O
(окисление йода до йодноватой кислоты пероксидом водорода),
2) 5H2
O2
+ 2HIO3
→ I2
+ 5O2
+ 6H2
O
(восстановление йодноватой кислоты до йода пероксидом водорода с выделением кислорода).
Брей обнаружил периодическое выделение кислорода из системы, зафиксировав несколько периодов сильно затухающих колебаний. Некоторые исследователи, ссылаясь на интенсивное выделение газа, высказывали сомнения в гомогенном характере этой реакции, поэтому существование колебательной реакции именно в гомогенной среде опыты Брея так и не доказали. В ряде своих последующих работ, однако, и сам Брей пытался доказать, что причиной колебаний являются гетерогенные стадии или процессы переноса.
И только в 1951 году советский химик Б. П. Белоусов открыл реакцию, в реальность которой нельзя было не поверить. В ходе реакции окисления лимонной кислоты броматом BrO3
-
, катализируемой ионами церия, он обнаружил регулярные периодические колебания окраски раствора от бесцветной (ионов Се3+
) к желтой (ионов Се4+
), и наоборот. Белоусов провел достаточно подробное исследование этой реакции и, в частности, показал, что период колебаний сильно уменьшается с повышением кислотности среды и температуры. Реакция весьма удобна для лабораторных исследований. Колебания можно было легко наблюдать визуально, а их период находился в пределах 10-100 секунд, совпадая с естественным масштабом времени наблюдателя-человека. Считается, что современная история исследований колебательных химических реакций в жидкой фазе началась в 1951 году именно с открытия реакции Белоусова-Жаботинского[14]
. Сходная реакция БриггсаРаушера, открытая в 1973 году, была названа йодными часами. Эта реакция похожа на реакцию Брея-Либавского и, кроме того, включает в себя некоторые элементы реакции Белоусова. В состав реакционной системы входят йодат калия KIO3
, пероксид водорода H2
O2
, хлорная HClO4
(или серная H2
SO4
) кислота, CH2
(COOH)2
, сульфат марганца MnSO4
, крахмал в качестве индикатора. В процессе этой реакции периодически изменяются концентрации йода I2
и йодид-ионов I-
, следить за колебательным процессом можно, наблюдая за появлением и исчезновением синей окраски, которую приобретает крахмал в присутствии йода.
11.2. Реакция Белоусова-Жаботинского
Реакция Белоусова-Жаботинского – это
протекающее в автоколебательном режиме каталитическое окисление различных восстановителей бромноватой кислотой НВrO3
. При этом наблюдаются колебания концентраций окисленной и восстановленной форм катализатора и некоторых промежуточных продуктов. Реакция идет в кислой среде, в водном растворе; в качестве катализаторов
используют ионы металлов переменной степени окисления, например церия, железа или марганца. В роли восстановителей выступают малоновая кислота, ацетилацетон и др.
Наиболее изящно, эстетически зрелищно выглядит колба, в которой идѐт реакция Белоусова-Жаботинского, если вместо лимонной кислоты использовать малоновую кислоту и ввести в систему раствор ферроина, комплекса железа(II) с индикатором о
-фенантролином. Общее уравнение реакции выглядит достаточно просто, однако реакция протекает более чем в 20 стадий и с образованием соответственно такого же количества промежуточных продуктов.
«Вы смотрите на стакан с красно-лиловой жидкостью, а она вдруг становится ярко-синей. А потом снова красно-лиловой. И снова синей. И вы невольно начинаете дышать в такт колебаниям. А когда жидкость налита тонким слоем, в ней распространяются волны изменения окраски. Образуются сложные узоры, круги, спирали, вихри, или все приобретает совершенно хаотический вид[15]
» – так описывает эту гомогенную колебательную химическую реакцию профессор С.Э.Шноль из Института химической физики АН СССР.
В реакции Белоусова-Жаботинского можно выделить три этапа:
Этап I
Br–
+ BrO3
–
+ 2 H+
→ HBrO2
+ HOBr
Br–
+ HBrO2
+ H+
→ 2 HOBr
Br–
+ HOBr + H+
→ Br2
+ H2
O
Br2
+ HOOCCH2
COOH→ HOOCCHBrCOOH + Br–
+ H+
Бромноватая кислота HBrOз
, реагируя с бромид-ионами (Br–
), образует бромноватистую кислоту HBrO2,
которая бромирует малоновую кислоту (HOOCCH2
COOH). В результате концентрация бромид-иона снижается.
Этап II
HBrO2
+ BrO3
–
+ H+
→ 2 .
BrO2
+ H2
O
.
BrO2
+ Fe++
+ H+
→ Fe+++
+ HBrO2
2 HBrO2
→ HOBr + BrO3
–
+ H+
Вследствие временного снижения концентрации бромид-иона HBrO2
реагирует с броматом (BrO3
–
) с образованием свободного радикала .
BrO2
[16]
. Этот свободный радикал окисляет Fe++
до Fe+++
(в присутствии которого окраска раствора становится красно-лиловой).
Этап III 2 Fe+++
+ 2 HOOCCHBrCOOH +
HOOCCH2
COOH + 3 H2
O
2 Br–
+ 2 Fe++
+ 3 HOOCCHOHCOOH + 4 H+
Броммалоновая кислота (образовавшаяся на Этапе I) восстанавливает Fe+++
до Fe++
(в присутствии которого раствор приобретает синюю окраску). При этом снова образуется бромид-ион и достигает концентрации, достаточной для того, чтобы инициировать новые реакции на Этапе I.
Рис.11.1 Узоры, которые образуются при выполнении реакции БелоусоваЖаботинского в тонком слое жидкости.
 
11.3. Колебательные реакции и синергетика
Объяснения колебательным химическим реакциям долгое время не было. Лишь во второй половине XIX в. возникли термодинамика и химическая кинетика, положившие начало специфическому интересу к колебательным реакциям и методам их анализа. В то же время развитие равновесной термодинамики послужило на первых порах тормозом при изучении подобных процессов. В реакции Белоусова-Жаботинского, например, в системе реагирующих частиц из хаоса сам собой возникает порядок: в какой-то момент все ионы железа (или церия) дружно переходят в одно состояние (Fe+3
или Ce+4
) и раствор приобретает синий цвет, однако система не остаѐтся в этом равновесном состоянии. Через строго определенное время все реагирующие частицы так же дружно переходят в новое состояние (Fe+2
или Ce+3
), и раствор становится красно-лиловым. И этот процесс так и продолжается с ритмической сменой цвета и идеально регулярным периодом колебаний. Однако самопроизвольное возникновение такой упорядоченности запрещает второй закон термодинамики
, так как в соответствии с ним в системе самопроизвольно может возрастать только хаос, или энтропия.
По словам профессора Шноля, «не мог образованный человек представить себе в беспорядочном тепловом движении огромного числа молекул макроскопическую упорядоченность, когда все молекулы дружно переходят то в одно, то в другое состояние! Это всѐ равно, что признать существование вечного двигателя. Этого быть не может!». На самом же деле системы, в которых идут колебательные процессы не нарушают второго закона термодинамики, потому что такие системы являются диссипативными
(от английского слова dissipate
– рассеивать). Система Белоусова-Жаботинского, двигаясь к состоянию равновесия, постоянно рассеивает энергию. В этом случае возможно временное присутствие некоторых промежуточных соединений в высоких концентрациях, в то время как другие промежуточные соединения присутствуют в низких концентрациях, и эта ситуация периодически меняется на противоположную. Единственным условием, которое второй закон термодинамики накладывает на эти локализованные концентрации, является требование, чтобы потеря энтропии в результате образования этих локализованных концентраций с избытком компенсировалась ростом энтропии в других процессах, которые одновременно происходят в процессе реакции.
Простейшая модель колебательной реакции математически тождественна уравнениям, которые итальянский ученый В. Вольтерра в начале 1930-х годов использовал для описания экологических процессов. В настоящее время это модель называется «моделью Лотка–Вольтерра» и описывает периодические изменения численности «жертвы» и «хищника» в экологических системах. Профессор Саратовского государственного университета С.П. Муштакова рассматривает колебательную реакцию как взаимодействие двух систем, одна из которых черпает необходимую ей для развития энергию, вещество или другие компоненты из другой. Такая задача называется задачей о хищниках и жертвах.
Представим, что в некоторой ограниченной экологической системе обитают волки и зайцы. В данной системе растет трава, которой питаются зайцы, в свою очередь являющиеся пищей для волков. В любой популяции при благоприятных условиях численность особей теоретически может неограниченно возрастать. В реальности же процесс роста ограничивается внешними факторами, например недостатком энергии или пищи. До определенного момента взаимодействие двух подсистем, т. е. популяций волков и зайцев, было сбалансированным: зайцев (с учетом их естественного пополнения) как раз хватало, чтобы прокормить определенное число волков. Затем в некоторый момент, принимаемый за нуль отсчета времени, из-за изменения какого-то фактора число зайцев возросло, что увеличило количество пищи для волков. Сытые волки начинают усиленно размножаться, давая новое потомство, которое на обильной пище быстро взрослеет и дает новый приплод. Складывается ситуация, когда зайцы уже не в состоянии прокормить всех волков, так как численность зайцев начинает падать, а волков (до поры) продолжает расти. Наконец экосистема перенаселена волками, а зайцев в ней становится всѐ меньше. Но тогда они становятся всѐ более трудной добычей для волков, и экосистема вступает в следующую фазу: численность зайцев уже упала до минимального уровня, при котором они практически неуловимы для волков. Поголовье волков, пройдя через максимум, начинает сокращаться, и это сокращение продолжается до тех пор, пока не будет достигнут такой его уровень, который в состоянии прокормить зайцы при своей минимальной численности. Теперь, когда численность волков достигла минимума, некому охотиться за зайцами. Зайцы начинают плодиться, а скудному волчьему поголовью за ними уже не уследить. Численность зайцев в короткий срок достигнет уровня, при котором они будут в состоянии прокормиться травой. Вновь возникает изобилие зайцев.
Сопоставляя схему «хищник-жертва» и колебательную реакцию, можно сделать вывод, что существуют факторы, без которых колебательный процесс был бы невозможен. Во-первых,
кооперативное поведение молекул в растворе невозможно без обратной связи
. Смысл обратной связи можно понять на примере взаимодействия зайцев и волков: увеличение числа особей хищника ведет к уменьшению популяции жертв, и наоборот. Наличие такой обратной связи обеспечивает устойчивое существование экосистемы. Если описывать колебательные химические реакции в терминах «хищник–жертва», то роль «хищников» выполняют промежуточные продукты, которые замедляют или совсем блокируют отдельные стадии процесса, – ингибиторы. Роль «жертв» выполняют катализаторы, которые ускоряют ход реакции. Хотя, как известно, катализатор (в реакции БЖ это церий или железо) не расходуется в реакции, но соотношение концентраций ионов [Fе2+
]/[Fе3+
], как показали исследования, претерпевает сложную эволюцию. Эта упрощенная схема позволяет в общих чертах представить молекулярный механизм обратной связи в растворе. Вовторых,
колебательный процесс невозможен без источника энергии
, роль которого в модели Лотка–Вольтерра выполняла трава, которую поедали зайцы. Очевидно, что ни о каких колебаниях и тем более устойчивости цикла «хищник–жертва» не может быть и речи, если в заповеднике забетонировать всю территорию – волки съедят зайцев и потом сами вымрут. В реакции Белоусова–Жаботинского источником энергии служит органическая малоновая кислота. При ее полном окислении колебания в реакции затухают, а затем и сама реакция прекращается.
Создатель теории неравновесных термодинамических процессов
, бельгийский учѐный русского происхождения И.Р.Пригожин
показал, что в открытой системе вблизи стационарного состояния, достаточно удаленного от химического равновесия, возможно возникновение порядка из хаоса, то есть самоорганизация системы
. При этом система должна рассеивать энтропию в окружающую среду, то есть быть диссипативной. Современная теория самоорганизации сложных систем называется синергетикой
(от греческого слова синергос
– совместное действие). Системы, способные к самоорганизации, развиваются по общим законам, независимо от природы систем. Колебательные химические реакции имеют особое значение для синергетики, так как их можно использовать в качестве достаточно простых моделей при изучении целого ряда сложных периодических процессов. Колебательный характер носят многие природные явления, от образования галактик до работы сердечной мышцы. Помогают также такие модели при разработке технологических процессов и даже новых материалов.
11.4. Практическое применение колебательных процессов
Интерес к колебательным системам постоянно растет, что свидетельствует о перспективности исследований в данном направлении. Одним из направлений прикладных исследований является изучение особенностей полимеризации в системе БелоусоваЖаботинского или в сходных с ней системах. Сложной пространственновременной организации, проявляемой системой БелоусоваЖаботинского (БЖ-системой) в отсутствие перемешивания, со временем нашлись аналогии в природе, в биологических системах (например, изучение фибрилляции сердечной мышцы с точки зрения рассмотрения миокарда как самоорганизующейся биологической системы). В 1974 г., профессором химии и биологии Аризонского университета (США)
А.Т.Уинфри были открыты пространственно-временные структуры в БЖ-системе без перемешивания, возникающие и существующие в виде различных двух- и трехмерных пространственных рисунков (например, концентрических колец, спиралей, волновых фронтов и т. п.). На Рис. 11.2. показаны образцы таких рисунков.
Недавно группе учѐных из Университета в Питтсбурге в США под руководством химика Анны Балац удалось смоделировать некий гелеподобный материал, который начинает пульсировать, вступая в реакцию с определѐнными химическими веществами. Благодаря этой особенности материала исследователи надеются, что в будущем смогут использовать его для приведения в движение миниатюрных роботов и прочих механических устройств.
Рис. 11.2. Так выглядят хаотические автоколебательные процессы в реакции Белоусова-Жаботинского.
 Впервые подобные гели были открыты в 1996 году сотрудниками японского Национального Института материалов и химикатов. Эти вещества, получившие название «гели Белоусова-Жаботинского» (так как механизм действия гелей сходен с механизмом реакции БЖ, см. Рис.11.2) состоят из длинных полимерных Впервые подобные гели были открыты в 1996 году сотрудниками японского Национального Института материалов и химикатов. Эти вещества, получившие название «гели Белоусова-Жаботинского» (так как механизм действия гелей сходен с механизмом реакции БЖ, см. Рис.11.2) состоят из длинных полимерных
молекулярных цепочек, а в качестве катализатора используют редкий металл рутений, по своей структуре весьма схожий с платиной. При добавлении в гель специальных азотистых соединений, в нѐм начинается колебательная химическая реакция, в результате которой рутениевый катализатор поочерѐдно то теряет, то притягивает электроны, заставляя тем самым полимерные цепочки то уменьшаться, то увеличиваться в длине. Это и приводит к пульсации вещества, которая, по словам исследователей, может длиться до нескольких часов. С помощью такого материала можно приводить в движение миниатюрных роботов, для которых он будет служить неким подобием искусственных мышц. Возможно, что впоследствии будет найден способ превращения этих пульсаций в электроэнергию, и на основе этого изобретения в будущем будет построен источник питания для ноутбуков, мобильных телефонов и прочей портативной техники.
Синергетический подход к изучению сложных и способных к самоорганизации систем привѐл к возникновению и развитию совершенно новой технологической отрасли – нанотехнологии.
11.5. Нанотехнологии
«Любой материальный предмет - это всего лишь скопление атомов в пространстве. То, как эти атомы собраны в структуру, определяет, что это будет за предмет», - вот основная идея, которая определяет назначение и возможности нанотехнологии. Из аналогии с существующими ныне микротехнологиями, оперирующими величинами порядка 0,1 - 1, 0 микрометра, или 10-6
м, следует, что нанотехнологии - это технологии, оперирующие величинами порядка нанометра, или 10-9
м. Это ничтожно малая величина, в сотни раз меньшая длины волны видимого света и сопоставимая с размерами атомов. Поэтому переход от «микро» к «нано» - это переход в новое качество. Оказалось, что природные и искусственные материалы имеют совершенно разные свойства на макро- и наноуровне. Так, наночастицы золота проявляют каталитические свойства, тогда как на макроуровне слиток золота — один из самых инертных материалов. Другие материалы имеют удивительные оптические свойства, например, сверхтонкие пленки органических веществ применяют для производства солнечных батарей. Такие батареи, хоть и обладают сравнительно низкой квантовой эффективностью, зато более дешевы и обладают ценными механическими свойствами, например, повышенной гибкостью. Система упорядоченных наночастиц часто проявляет необычные свойства, главным из которых является то, что сверхчистые наночастицы могут образовывать упорядоченные структуры, то есть обладают способностью к самоорганизации
.
Удается добиться взаимодействия искусственных наночастиц с природными объектами наноразмеров — белками, нуклеиновыми кислотами, что открывает захватывающие перспективы для развития нанотехнологий
. Нанотехнология, или молекулярная нанотехнология, или МНТ, - это область науки и техники, связанная с разработкой нанороботов
, устройств размером порядка нанометра, состоящих из атомов, количество которых может колебаться от нескольких десятков до нескольких сотен тысяч. Основное назначение таких устройств – оперирование отдельными атомами и молекулами на межатомных расстояниях, которые измеряются десятыми долями нанометра.
Импульс развитию нанотехнологии дало создание сканирующих туннельных микроскопов (scanning-tunneling microscope) СТМ или атомных силовых микроскопов (atomic-force microscope) АФМ, которые позволяют исследовать вещество на атомном уровне, то есть видеть отдельные атомы и перемещать их в пространстве (См. Рис. 12.3). За это изобретение в 1986 году группе американских разработчиков была присуждена Нобелевская премия. Нанотехнологии развиваются в трех направлениях:
- изготовление наноэлектронных схем
(в том числе и объемных) с активными элементами, размерами сравнимыми с размерами молекул и атомов; при этом можно воспроизвести нейронные интегральные схемы для компьютеров, на которые возлагаются большие надежды. В то же время, сейчас активно развиваются нанотехнологические методы, позволяющие создавать активные элементы (транзисторы, диоды) размером с молекулу и формировать из них многослойные трехмерные схемы.
- разработка и изготовление наномашин
, т.е. механизмов и роботов размером с молекулу. Они будут снабжены миниатюрным вычислительным устройством и манипуляторами, позволяющими работать с молекулами - например, перемещать их и модифицировать их структуру, т.е. заниматься молекулярной хирургией. Аналогом простейшего молекулярного робота является рибосома, строящая из аминокислот молекулу белка по «программе», которой является молекула транспортной рибонуклеиновой кислоты (см. Лекцию 18).
- непосредственная манипуляция атомами
и молекулами и сборка из них всего существующего. Нанотехнологии в будущем, возможно, будут использовать, чтобы вернуть человеку здоровье, потому что человеческое тело состоит из атомов и молекул. Больные и старые люди страдают оттого, что молекулы, из которых состоят их тела, складываются в неправильные структуры вследствие вторжения в организм вирусов, старения клеток организма или травм. Устройства, способные переупорядочить атомы, смогут переустанавливать их в правильное положение. Нанотехнология приведет к фундаментальному прорыву в медицине.
Первые результаты по перемещению единичных атомов и сборки из них определенных конструкций появились ещѐ в конце XX века. Тогда же были разработаны и изготовлены первые наноэлектронные элементы. В настоящее время нанотехнологии стремительно развиваются, в частности, для производства наноэлектронных чипов и микросхем памяти емкостью в десятки гигабайт. Более отдаленные перспективы применения нанотехнологий сейчас даже трудно себе вообразить.
Одним из важнейших вопросов, стоящих перед нанотехнологией — как заставить молекулы группироваться определенным способом, самоорганизовываться, чтобы в итоге получить новые материалы или устройства. Этой проблемой занимается особый раздел химии — супрамолекулярная, или когерентная химия
. Она изучает не отдельные молекулы, а взаимодействия между молекулами, которые, самоорганизовываясь определенным способом, могут образовывать сложные наносистемы, например, наномашины. Обнадеживает то, что в природе действительно существуют подобные системы и осуществляются подобные процессы, например, рассмотренные в данной лекции колебательные процессы, в частности, реакция Белоусова-Жаботинского.
Рис. 11.3. Сканирующий туннельный микроскоп позволяет манипулировать объектами наноразмеров

11.6. Вопросы и задания
1. Что такое «кольца Лизеганга»?
2. Как протекают реакции Брея-Либавского и Бриггса-Раушера?
3. Опишите реакцию Белоусова-Жаботинского. Почему, наблюдая за этим процессом, химик, убеждѐнный в справедливости второго закона термодинамики, восклицает: «Этого не может быть!»?
4. Опишите механизм реакции БЖ на основе схемы «хищник-жертва».
5. Что такое диссипативные системы?
6. Что изучает синергетика?
7. Дайте характеристику системам, способным к самоорганизации.
8. Каково практическое применение колебательных процессов?
9. Что такое нанотехнологии?
10. Что такое супрамолекулярная, или когерентная, химия?
Лекция 12. Реакционная способность веществ
При определенных условиях каждая химическая реакция самопроизвольно протекает в прямом или обратном направлении.
Например, при низких температурах процесс:
 Н2(г)
+ О2(г)
= 2Н2
О(г)
( Н = -241,8 кДж/моль) Н2(г)
+ О2(г)
= 2Н2
О(г)
( Н = -241,8 кДж/моль)
протекает хотя и очень медленно, но целиком в прямом направлении с образованием паров воды. Однако при высоких температурах эта реакция начинает идти в обратном направлении: водяной пар разлагается на водород и кислород. В чем же причина определенной направленности химических процессов, какие факторы на эту направленность влияют? Эта проблема называется проблемой химического сродства
.
12.1. Химическое сродство
Химическим сродством называют способность компонентов системы вступать в химическую реакцию друг с другом. Чтобы решить проблему химического сродства, необходимо установить, какие факторы оказывают влияние на поведение химических веществ, так что одни вещества охотно (самопроизвольно) вступают в реакцию друг с другом, а другие реагируют только в экстремальных условиях. Необходимо также определить количественную меру химического сродства
.
Любые элементы любой системы (например, электроны в атоме или два атома, образующие молекулу) стремятся занять такое положение, чтобы их энергия была минимальной. Этот принцип называется принципом минимальной энергии
. Естественно предположить, что и химические процессы должны самопроизвольно протекать в направлении уменьшения внутренней энергии системы, то есть система в ходе химической реакции должна стремиться избавиться от «лишней» энергии, выбросив ее в окружающую среду в виде энтальпийной теплоты.
Однако, для того, чтобы сказать, когда реакция может идти самопроизвольно, например, в стандартных условиях (под стандартными условиями понимают такие условия, когда температура равна 298 градусам Кельвина, а давление – одной атмосфере, или 101 кПа), недостаточно знать только ее энтальпию. Если бы был справедлив только принцип минимальной энергии, самопроизвольно при нормальных условиях шли бы только экзотермические реакции. Однако опыт показывает, что это не так. Что же еще нужно принимать в расчет?
Необходимо вспомнить о втором законе термодинамики
(см. Лекцию 7, раздел 7.2), который утверждает, что самопроизвольно происходит только процесс перехода от порядка к беспорядку, или от меньшего беспорядка к большему беспорядку, потому что состояние беспорядка более вероятно, чем упорядоченное состояние. Именно это и должно происходить в любой колбе, автоклаве или ректификационной колонне, так как согласно со вторым началом термодинамики энтропия в изолированной системе стремится к максимуму.
Рассмотрим несколько реакций и попробуем предсказать, в каком направлении они пойдут стандартных условиях:
 1) H2(г)
↔ 2H(г)
;
H
+463 кДж/моль 1) H2(г)
↔ 2H(г)
;
H
+463 кДж/моль
2) H2
O (ж)
+ SO3 (г)
↔ H2
SO4 (ж)
; H
- 217 кДж/моль 3) CH4
(г)
+2O2(г)
↔ CO2(г)
+ 2H2
O(г)
; H
-890,3 Кдж/моль 4) 2C3
H5
N3
O9 (ж)
↔
6CO2(г)
+5H2
O(г) + 3N2(г)
+ ½ 02(г)
; H
-1430 кДж/моль
 5) 2H2(г)
+ O2(г)
↔ 2H2
O(ж)
;
H
-285,8 кДж/моль 5) 2H2(г)
+ O2(г)
↔ 2H2
O(ж)
;
H
-285,8 кДж/моль
В реакциях 1 и 4 несомненна большая беспорядочность у продуктов реакции, в результате реакций 2 и 5 беспорядок уменьшается, а о реакции 3 ничего определенного сказать нельзя – число молекул в левой и правой части одинаково. Основываясь на энтропийном факторе
изложенных выше рассуждений можно было бы сказать, что самопроизвольно будут происходить только прямые реакции 1 и 4, в которых направление хода прямой реакции совпадает с направлением «порядок → беспорядок». Для реакций же 2 и 5 можно было бы ожидать самопроизвольного их протекания в обратном порядке
(самопроизвольно пойдѐт обратная реакция).
Наши рассуждения не согласуются с опытом: если смешать вышеназванные реагенты в стандартных условиях, то реакции 2 – 5 пойдут в прямом направлении (слева направо), а реакция 1 пойдет справа налево, то есть в обратном направлении. Мы не приняли в расчет энтальпийный фактор
: взаимодействие серного ангидрида с водой, сжигание метана, разложение нитроглицерина и образование воды – все эти превращения сопровождаются большим выделением энергии. В случае разложения нитроглицерина стремление системы к неупорядоченности и стремление ее же избавиться от «лишней» энергии совпадают – и реакция происходит в виде взрыва. В реакции 3 не происходит значительного увеличения беспорядка – и самопроизвольный ход реакции определяет, в основном, энтальпийный фактор: выделение 890,3 кДж/моль теплоты при окислении метана позволяет системе «сбросить» лишнюю энергию. В реакциях 2 и 5 выделение энергии в ходе прямой реакции компенсирует невыгодность перехода в боле упорядоченное состояние. А вот в реакции 1 нужно затратить 463 кДж на достижение большей неупорядоченности. Такое состояние стоит слишком дорого, и реакция идет в обратном направлении – от атомов водорода к молекуле Н2
. Следовательно, делая заключение о возможности или невозможности самопроизвольного протекания реакции нужно принимать в расчет два фактора: тепловой эффект, или энтальпию реакции, и стремление системы к беспорядку, то есть энтропийный фактор. Энтропия системы растѐт с повышением температуры, следовательно, температуру необходимо обязательно принимать в расчѐт.
12.2. Энергия Гиббса
В химических процессах одновременно действуют два противоположных фактора — энтропийный (TΔS) и энтальпийный (ΔH). Суммарный эффект этих противоположных факторов в процессах, протекающих при постоянном давлении и температуре называется изменением энергии Гиббса[17]
(G
):
 G H T S
(12.1) G H T S
(12.1)
Из этого выражения следует, что ΔH
= ΔG
+ T
ΔS
, то есть некоторое количество теплоты расходуется на увеличении энтропии (T
ΔS
). Эта часть энергии потеряна для совершения полезной работы, еѐ часто называют связанной энергией
. Другая часть теплоты (ΔG
) может быть использована для совершения работы, поэтому энергию Гиббса часто называют также свободной энергией
. Характер изменения энергии Гиббса позволяет судить о принципиальной возможности осуществления процесса. Если свободная энергия Гиббса в исходном состоянии системы больше, чем в конечном состоянии (то есть если G1
больше G2
, а G
<0), то процесс принципиально может протекать, если наоборот (то есть если G1
меньше G2
, а G
>0), — то не может.
Или:
При ΔG
< 0 самопроизвольный процесс возможен;
При ΔG
> 0 процесс самопроизвольно протекать не может; Если же ΔG
= 0, то система находится в состоянии химического равновесия.
Самопроизвольно могут протекать только те реакции, в которых свободная энергия Гиббса уменьшается. Если бы такое соотношение между величинами свободной энергии сохранялось на протяжении всей реакции, то процесс шел бы до конца, до полного исчезновения всех исходных веществ. Но на деле все происходит иначе. В ходе химической реакции величина свободной энергии исходных веществ падает, а величина свободной энергии продуктов реакции растет. Через некоторое время величины свободной энергии сравняются. В этом состоянии у реакции нет никакого стимула идти ни слева направо, ни справа налево. Такое состояние может сохраняться неограниченно долго и называется
состоянием равновесия. Это понятие применимо не только к химическим системам, но здесь оно наиболее наглядно. Оно отражает очевидный факт:
система, находящаяся в состоянии равновесия, не может производить работу. Состояние равновесия наступает, когда изменение свободной энергии Гиббса равно нулю. Изменение свободной энергии Гиббса является количественной мерой химического сродства, или критерием самопроизвольности протекания химического процесса, имеющего место в химической системе с неизменной температурой и давлением. Такие процессы называются избарно-изотремическими. Поэтому энергию Гиббса называют ещѐ
изобарно-изотремическим потенциалом.
12.3. Энергия Гельмгольца
 Для реакции, протекающей при постоянной температуре и постоянном объѐме, мерой химического сродства является изменение изохорноизотермического потенциала, или свободной энергии Гельмгольца
F
. Свободная энергия Гельмгольца связана с внутренней энергией U и энтропией S соотношением: Для реакции, протекающей при постоянной температуре и постоянном объѐме, мерой химического сродства является изменение изохорноизотермического потенциала, или свободной энергии Гельмгольца
F
. Свободная энергия Гельмгольца связана с внутренней энергией U и энтропией S соотношением:
 F U T S (12.2) F U T S (12.2)
Внутренняя энергия связана с энтальпией соотношением:
 U H nRT (12.3) U H nRT (12.3)
где R – универсальная газовая постоянная (8,3144103
Дж/кмольК), а
 n - изменение числа молей вещества в реакции. n - изменение числа молей вещества в реакции.
Следовательно, энергия Гиббса и энергия Гельмгольца связаны друг с другом соотношением:
 F U G (12.4) F U G (12.4)
Самопроизвольная реакция в системе с постоянным давлением и температурой возможна, если F
< 0
Пусть, например, нужно вычислить энтальпию, изменение энергии Гиббса и Гельмгольца, а также полную энергию для следующей
реакции: СО2(г)
+4Н2(г)
↔СН4(г)
+2Н2
О(г)
и определить, в каком направлении пойдет реакция при стандартном давлении и температуре (298 К).
Соответствующие термодинамические величины приведены в нижеследующей таблице.
Таблица 12.1. Термодинамические величины
Вещество H 0
298
кДж/моль G 0
298
кДж/моль
| Н2О(г)
|
-241,84
|
-228,8
|
| CН4(г)
|
-74,85
|
-50,79
|
| СО2(г)
Решение:
|
-393,51
|
-394,38
|
Энтальпию данной химической реакции Hнаходим из следствия из закона Гесса как разность между суммами теплот образования продуктов реакции и исходных веществ, или
 H 0298 х.р. = 2 H 0298 Н2О(г) + H 0298 CН4(г) - 4 H 0298 H2(г) - H 0298 СО2(г) H 0298 х.р. = 2 H 0298 Н2О(г) + H 0298 CН4(г) - 4 H 0298 H2(г) - H 0298 СО2(г)
Теплоты образования соответствующих веществ находим из таблицы термодинамических величин, учитывая, что теплота образования простого вещества принимается равной нулю. Тогда:
H 0
298
х.р.
= 2∙ (-241,84) + (-74,85)- 4∙0 – (-393,51) = -165,02 кДж/моль
 Аналогично вычисляем изменение энергии Гиббса G(в стандартных условиях): Аналогично вычисляем изменение энергии Гиббса G(в стандартных условиях):
G 0298х.р. = G 0298 Н2О(г) + G 0298 CН4 -4 G 0298H2 - G 0298 СО2 ; значения свободных энергий образования интересующих нас веществ находим из таблицы термодинамических величин. Тогда:
G0
298
х.р.
= 2(-228,8) + (-50,79) - 4∙0 – (-394,38) = -114,01 кДж/моль
Поскольку изменение энергии Гиббса меньше нуля, в стандартных условиях реакция протекает в прямом направлении.
Изменение полной энергии системы находим из соотношения:
U H nRT; где R = 8,31∙10-3
кДж/моль∙К, а n (изменение количества молей вещества в ходе реакции) = 3-5 = -2 Отсюда:
 U
165,02 ( 2)8,31 10 3
298 = -160,067 кДж/моль U
165,02 ( 2)8,31 10 3
298 = -160,067 кДж/моль
Свободная энергия Гельмгольца определяется как разность
  F U T S, или F U G= -160,067 – (-114,01) = -46,06 кДж/моль F U T S, или F U G= -160,067 – (-114,01) = -46,06 кДж/моль
12.4. Критерий возможности самопроизвольного протекания химических процессов
Для удобства сравнения различных реакций принято сравнивать значения свободной энергии Гиббса при стандартных условиях. Термодинамические величины, характеризующие вещество в его стандартном состоянии, называются стандартными величинами. Стандартные величины и их изменения принять обозначать при помощи знака «0
298
». Например, стандартное изменение энтропии обозначается
S
298
0
, стандартное изменение энтальпии H
298
0
, а стандартное изменение энергии Гиббса G
298
0
. Стандартным состоянием вещества называется
(см. Лекцию 7) его состояние при температуре 298,16 градусов К (25о
С) и давлении 1 атмосфера (101 кПа).
Для вычисления стандартных изменений энергии Гиббса обычно используют энергии Гиббса образования сложных химических веществ из простых веществ (при стандартных условиях). Энергия Гиббса образования простых веществ принимается равной нулю
. По следствию из закона Гесса (см. Лекцию 9), стандартное изменение энтальпии реакции равно сумме стандартных энтальпий образования продуктов реакции за вычетом суммы стандартных энтальпий образования исходных веществ.
Аналогично, стандартное изменение энергии Гиббса реакции равно сумме стандартных энергий Гиббса образования продуктов реакции за вычетом суммы стандартных энергий Гиббса образования исходных веществ.
 (12.5) (12.5)
А также, стандартное изменение энтропии реакции равно сумме стандартных энтропий образования продуктов реакции за вычетом суммы стандартных энтропий образования исходных веществ.
 (12.6) (12.6)
Для того, чтобы определить, может ли самопроизвольно протекать при стандартной температуре 289,160К реакция:
Na2
O(к) + Н2
О(ж) = 2NaOH(к)
рассчитаем стандартную энергию Гиббса, исходя из табличных значений стандартных энергий Гиббса образования исходных веществ и продуктов реакции и соотношения 12.5.
Таблица 12.2. Термодинамические величины
| Вещество
|
ΔН0298 кДж/моль
|
ΔS0298
Дж/моль∙К
|
ΔG0298 кДж/моль
|
| Na2
O(к)
|
-430,6
|
71,1
|
- 376,6
|
| Н2
О(ж)
|
-285,84
|
69,96
|
- 377,0
|
| NaOH(к)
|
-426,6
|
64,18
|
- 273,5
|
ΔG0298 = 2ΔG0298 образования NaOH(к) - ΔG0298 обр. Na2O(к) - ΔG0298 обр. Н2О(ж) ; ΔG0
298
= 2(-377, 0) – (-376,0) – (-237,5) = -140,5 кДж/моль.
Так как ΔG0
298
меньше нуля, процесс может идти самопроизвольно при 2980
К и давлении 101кПа.
Если реакция протекает не в стандартных условиях, для расчѐта изменения свободной энергии Гиббса следует пользоваться соотношением (12.1). Выясним, идѐт ли реакция 12.4 самопроизвольно при температуре 100о
С (398 К).
Используя данные таблицы термодинамических величин и соотношение
(9.8), находим изменение энтальпии данной реакции:
H
= 2(-426,6) – (-430,6) – (-285,84) = -136,76 кДж/моль.
Находим изменение энтропии в реакции, пользуясь соотношением (12.6) и данными таблицы:
S
= 2∙64,18–71,1- 69,96 = -12,7 Дж/моль∙К
Находим изменение свободной энергии Гиббса:
 G H T S
= -136,76 кДж/моль -298, 16К∙(-0,0127 кДж/моль∙К) = G H T S
= -136,76 кДж/моль -298, 16К∙(-0,0127 кДж/моль∙К) =
-140,5кДж/моль
Константа равновесия и стандартное изменение энергии Гиббса связаны соотношением:
  G
х
.р
.
2,3RT
lg K
, или G
х
.р
.
0,01915T
lg K
(12.7) G
х
.р
.
2,3RT
lg K
, или G
х
.р
.
0,01915T
lg K
(12.7)
Например, чтобы вычислить константу равновесия реакции
NH3(г) + HCl(г) = NH4Cl(к)
находим ΔG0
298
реакции, исходя из табличных значений свободных энергий образования исходных веществ и продуктов реакции: ΔG0
298
аммиака равно –16,7 кДж/моль; ΔG0
298
хлористого водорода равно –94,8 кДж/моль; ΔG0
298
хлористого аммония равно –203,2 кДж/моль. Поэтому: ΔG0
298
= -203,2 – (-16,7- 94,8) = -91,7 кДж/моль.
Подставляем найденное значение в уравнение (12.8) и получаем:
-91,7 = -5,71 lg K298
; lg K298
= 16; К298
= 1016.
Большая величина константы равновесия показывает, что при стандартных условиях равновесие реакции сильно смещено вправо, то есть, что хлористый аммоний при этих условиях – очень устойчивое соединение.
В электрохимических системах (см. Лекцию 4) критерием возможности самопроизвольного протекания электрохимической (окислительно-восстановительной) реакции тоже является отрицательное значение G токообразующей реакции. Так как 1 джоуль соответствует энергии заряда в 1 кулон, прошедшего разность потенциалов 1 вольт, а 1 моль зарядов соответствует числу Фарадея F, которое равно 96487 Кл (округлѐнно 96500 Кл). Тогда для изменения энергии Гиббса получаем соотношение:
G nFE
(12.8)
где n – число электронов, участвующих в токообразующей реакции; F – число Фарадея, а E – электродвижущая сила (ЭДС), или напряжение источника тока в отсутствие внешней нагрузки.
12.5. Вопросы и задания
1. Какие два фактора определяют возможность самопроизвольного протекания химических процессов?
2. Что такое энергия Гиббса?
3. Как связаны энергия Гиббса и энергия Гельмгольца?
4. Сформулируйте условие самопроизвольного протекания химических процессов.
5. Исходя из значений стандартных теплот образования и стандартных абсолютных энтропий соответствующих веществ, вычислите изменение энергии Гиббса ΔG0
298
реакции, протекающей по уравнению: NH3(г)
+ HCl(г)
 NH4
Cl(к)
. Может ли эта реакция идти самопроизвольно в стандартных условиях? Стандартные теплоты образования (энтальпии) газообразного аммиака, хлористого водорода и кристаллического хлорида аммония равны соответственно –46,19; -92,31 и –315,39 кДж/моль. Стандартные абсолютные энтропии газообразных аммиака, хлористого водорода и кристаллического хлорида аммония равны соответственно 192,50; 186,68 и 94,5 Дж/моль·К NH4
Cl(к)
. Может ли эта реакция идти самопроизвольно в стандартных условиях? Стандартные теплоты образования (энтальпии) газообразного аммиака, хлористого водорода и кристаллического хлорида аммония равны соответственно –46,19; -92,31 и –315,39 кДж/моль. Стандартные абсолютные энтропии газообразных аммиака, хлористого водорода и кристаллического хлорида аммония равны соответственно 192,50; 186,68 и 94,5 Дж/моль·К
6. Могут ли в стандартных условиях протекать следующие реакции?
CuO(к) + H2(г) ↔ Cu(к) + H2O(г)
ZnS(к) + O2(г) ↔ ZnO(к) + SO2(г)
CH4(г) + 2O2(г) ↔ 2H20(ж) + CO2(г)
SO2(г) + 2H2(г) ↔ 2S(к) + 2H2O(г)
4HCl(г) + O2(г) ↔ 2Cl2(г) + 2H2O(ж)
Стандартные изменения энергии Гиббса образования соответствующих веществ равны (кДж/моль):
ΔG0298 СuO(к) =-127,19; ΔG0298 SO2 (г) =-300,37; ΔG0298 CO2 (г) =-394,38
ΔG0298 H20(г) =-228,8; ΔG0298 CH4 (г) =-50,79; ΔG0298 ZnS(к) =-198,32
ΔG0
298
H2
O(ж)
= 237,5; ΔG0
298
HCl(г)
=-95,27; ΔG0
298
ZnO(к)
= -318,19
7. При какой температуре наступит равновесие системы: 4HCl(г)
+ O2(г)
↔ 2Cl2
(г)
+ 2H2O(ж)?
Стандартные энтальпии и энтропии образования соответственно равны: ΔH0
298
HCl(г)
=-92,30 кДж/моль ; ΔH0
298 H2O(ж)
=-285,84 кДж/моль; ΔH0
298 H2O(ж)
=-285,84 кДж/моль; ΔS0298 HCl(г) = 186,7дж/моль∙К; ΔS0298Cl2(г) = 223,0 Дж/моль∙К; ΔS0298 O2
(г
) = 205,03 Дж/моль∙К; ΔS0298H2O(ж)=69,96 Дж/моль∙К
8. Реакция CO + H2
O ↔ CO2
+ H2
протекает при 970 К и Р = 1,0133∙105
Па. По изменению энергии Гиббса определите, в какую сторону пойдет реакция, если исходная реакционная смесь имеет следующий состав (в молярных долях):
45% СО, 15% Н2
О, 25%СО2
и 15% Н2
.
9. Можно ли осуществить в гальваническом элементе следующую реакцию: Fe0
+ Cd2+
→ Fe2+
+ Cdo
? Стандартные электродные потенциалы железа и кадмия равны, соответственно, -0,44 В и -0,40 В.
10. Вычислите стандартную энергию Гиббса G 0
298
, стандартную энергию
Гельмгольца F298
0
и полную энергию U
для реакции HCl(г)= H2
(г) +Cl2(г)
.
Определите, в какую сторону пойдет реакция в стандартных условиях, если стандартная энтальпия образования хлористого водорода равна -92,30 кДж/моль, а стандартное изменение энергии Гиббса образования HCl равно -
95,27 кДж/моль.
Лекция 13. Химия и периодическая система элементов
Сегодня в системе Менделеева 117 элементов. У каждого элемента есть свое название и условное обозначение, состоящее из одной или двух латинских букв. Названия элементов складывались исторически. Так, например, название водород произошло от слов «рождающий воду» (hydrogenium), и первая буква латинского названия этого элемента стала его условным обозначением. Элемент фосфор обязан своему названию латинскому слову «phosphorus», что значит «светящийся». Элемент рутений назван в честь России. Многие элементы названы в честь выдающихся ученых, например кюрий или нильсборий. В природе в значительных количествах встречается всего около 90 элементов системы Менделеева. Элементы, стоящие в конце таблицы, все искусственного происхождения, их получают посредством ядерных реакций в ускорителях в количестве всего одного - двух атомов. Никакого практического применения, во всяком случае, в настоящее время, эти элементы не имеют. В последнее десятилетие в ускорителях элементарных частиц при ядерных реакциях получены следующие элементы: 104 – рутерфордий (курчатовий), 105 – дубний; 106 - сиборгий; 107 – борий, 108 – гассий, 109 – мейтнерий, 110 – унуннилий (дармштадтий), 111 – унунуний (рентгений), 112 – унунбий, 113 - унунтрий,114 – унунквадий, 115 – унунпентий, 116 – унунгексий, 117 – унунсептий, 118 – унуноктий. Условные названия элементам, начиная со 110, даются по номерам, при этом цифры номера называются по порядку по латыни. В скобках даны ещѐ не установившиеся названия новых элементов. Элемент с номером 117 до сих пор получить не удалось.
Сегодня существует около 400 разновидностей Периодической таблицы. Самой распространенной является восьмиклеточная таблица в короткопериодном варианте
, однако Международный союз чистой и прикладной химии (ИЮПАК) рекомендует использовать длиннопериодный вариант Периодической системы (см. Рис.13.1), в которой по рекомендации ИЮПАК вертикальные ряды (группы) Периодической системы нумеруются от №1 (щелочные элементы) до №18 (инертные газы).
Рис. 13.1. Длиннопериодный вариант Периодической системы элементов
Период
2
He
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 111
Rg
|
112
Uub
|
113 Uut
|
114
Uuq
|
115
Uup
|
116
Uuh
|
117
Uus
|
118
Uuo
|
|
| *Лантаниды
|
*
|
57
La
|
58
Ce
|
59 Pr
|
|
|
|
|
|
|
|
|
|
| **Актиниды
|
**
|
89
Ac
|
90
Th
|
91
Pa
|
|
|
|
|
|
|
|
|
|
13.1. Связь Периодической системы с квантовой химией
Периодический закон сыграл решающую роль в выяснении сложной структуры атома. Более того, обнаружилось, что система Менделеева основана на законах квантовой механики, чего не мог предвидеть даже сам ее автор. Связь Периодического закона с квантовой химией
состоит в следующем:
Порядковый номер элемента численно равен количеству протонов в ядре атома, или заряду атомного ядра. Атомы нейтральны, то есть положительный заряд ядра уравновешивается отрицательным зарядом электронов, число которых в нейтральном атоме также равно
порядковому номеру элемента; Массовое число (относительная атомная масса элемента) равно сумме протонов и нейтронов в ядре атома данного элемента;
Номер периода, в котором расположен элемент, указывает на то, на каких орбиталях с наибольшей вероятностью располагаются электроны в его атомах, то есть позволяет определить значения главного квантового числа n. Для всех элементов первого периода n = 1; для всех элементов второго периода n = 1, 2; для всех элементов третьего периода n = 1, 2, 3 и т.п. Зная главное квантовое число, можно определить и остальные квантовые числа любого электрона в данном атоме;
Номер группы (в 8-клеточной таблице) равен количеству электронов на внешнем электронном уровне атома данного элемента;
Пользуясь Периодической системой Менделеева и применяя законы квантовой механики, можно узнать, сколько неспаренных электронов имеется на внешнем электронном уровне атома данного элемента, а именно этим и определяются химические свойства элемента.
13.2. Строение Периодической системы
Периодическая система состоит из периодов и групп. Периодов в системе имеется семь, из них три малых и четыре больших. Номер периода соответствует числу главных энергетических уровней любого атома, расположенного в периоде (N = n). Главный уровень делится на s-, p-, f- и d- подуровни. Руководствуясь принципом Паули, можно подсчитать, какое максимальное количество электронов может расположиться на каждом подуровне. Для s -подуровня это число равно 2, для p - подуровня 6, для f - и d - подуровней - 10 и 14 соответственно.
Спаренные электроны, располагающиеся на заполненных в соответствии с принципом Паули и правилом Гунда орбиталях, как правило, не склонны покидать свои места, так как в спаренном состоянии энергия электронов меньше, чем в распаренном положении. Поэтому для химика наиболее важным является внешний электронный слой
атома, не заполненный до конца, потому что именно неспаренные электроны внешнего слоя образуют химические связи при взаимодействии атомов друг с другом.
Первый период состоит из двух элементов - водорода и гелия. В атоме водорода единственный электрон располагается в первом электронном слое 1s. В соответствии с принципом Паули, на одной орбитали могут находиться электроны с разными спинами, поэтому в атоме гелия два электрона расположены также на уровне 1s. Такая структура из двух спаренных электронов является энергетически устойчивой, поэтому, в отличие от химически активного водорода, гелий не вступает в химические реакции с другими элементами (не обменивается с ними электронами) и поэтому называется инертным газом
.
Во втором и третьем периодах расположено по 8 элементов. У элементов второго периода заполняется второй электронный слой (n=2), сначала 2s, а затем 2р - орбиталь, причем количество электронов на втором уровне постепенно возрастает от 1 до 8. Восьмиэлектронная оболочка (октет) неона представляет собой очень устойчивую энергетическую структуру, поэтому неон также является инертным газом. У элементов третьего периода заполняется третий электронный слой (у двух первых элементов заполняются 3s - орбитали, а у шести последних - 3р-орбитали). В отличие от второго периода, у элементов третьего периода остаются свободными 3d - орбитали, что также оказывает влияние на их химические свойства и свойства их соединений. Элементы, в атомах которых s - подуровень заполняется в последнюю очередь, называются s - элементами
; те элементы, в атомах которых последними заполняются р - подуровни, называются р- элементами
; соответственно определяется и отношение атомов к семействам d - и f - элементов.
Большие периоды четвертый и пятый содержат по 18 элементов. У элементов четвертого периода после заполнения 4s-слоя, - он заполняется раньше слоя 3d, из-за экранирования (заслонения) ядра плотным и симметричным слоем электронов на уровнях 3s и 3p, - начинает заполняться слой 3d. На этом подуровне может разместиться максимум 10 электронов, поэтому заполнение слоя 4р начинается только у галлия. У хрома и меди имеет место «провал» 4s - электрона на подуровень 3d. Таким образом, четвертый период начинается двумя s элементами и заканчивается шестью р - элементами, а между s- и pэлементами располагаются 10 переходных d -элементов.
В пятом периоде заполнение электронных уровней и подуровней (слоев и подслоев) происходит так же, как и в четвертом периоде: у двух первых элементов, рубидия и стронция, заполняется внешний 5s - слой, у шести последних (от индия до ксенона) - внешний 5d - слой. Между ними располагаются 10 d - элементов, у которых заполняется 4d – подслой, причѐм, у ниобия, молибдена, рутения, родия и палладия происходит «провал» электрона с уровня 5s на 4d.
Шестой период состоит из 32 элементов. Он тоже начинается двумя s-элементами (заполняется 6s-подуровень) и заканчивается шестью p-элементами (заполняется 6р-подуровень). У лантана начинает заполняться 5d-подуровень, а у следующих за лантаном четырнадцати элементов (их называют лантаноидами, или лантанидами, и выделяют в таблице отдельной строкой) заполняется 4f–подуровень - третий снаружи квантовый слой. Затем у элементов от гафния до ртути идет заполнение 5d-подуровня (всего 10 элементов), а у элементов от таллия до радона заполняется 6p-подуровень. Таким образом, в шестом периоде содержится два s - элемента, шесть p - элементов, десять d - элементов и четырнадцать f - элементов.
В седьмом периоде имеются два s - элемента (франций и радий), d-элемент актиний и следующие за ним четырнадцать f - элементов, называемых актиноидами, или актинидами (от тория до лоуренсия), далее опять следуют d-элементы (от элемента № 104, резерфордия, до элемента №118). Седьмой период ещѐ не завершен, так как получить элемент № 117 пока не удалось.
Таким образом, характерные для периодической системы
(«магические») числа - 2, 8, 18 и 32 - с необходимостью вытекают из теории строения атомов. Эти числа равны максимальному количеству электронов, могущих разместиться на s (2), s+p 2+6), s+p+d (2+6+10) и s+p+d+f (2+6+10+14) - электронных подуровнях.
Период представляет собой последовательный ряд элементов, в атомах которых происходит заполнение одинакового числа электронных слоев
. Разная длина периодов объясняется последовательностью заполнения электронных слоев (у s и p элементов заполняется внешний слой, у d- элементов предвнешний слой, а у f- элементов - третий снаружи). Поэтому отличия в свойствах наиболее отчетливо проявляются у s- и p- элементов, а различие химических свойств у d - и f -элементов одного и того же периода выражено менее резко. Последние элементы в пределах периода объединяются в семейства. Это семейства скандия, иттрия, гафния (d- элементы), а также лантаниды и актиниды (f - элементы).
Длиннопериодный вариант Периодической системы содержит 18 групп. В I и II группах содержатся s-элементы. Это активные щелочные и щелочноземельные металлы. У р-элементов XIV – XVII групп преобладают неметаллические свойства. Все р-элементы XVIII группы являются инертными газами, так как не имеют неспаренных электронов В группах с III по XII содержатся d-элементы, которые часто называют переходными, так как они проявляют как свойства металлов, так и свойства неметаллов. Все свойства элементов, определяемые строением электронной оболочки атома, закономерно изменяются по периодам и группам Периодической системы. При этом, поскольку электронные структуры элементов-аналогов сходны, но не тождественны, при переходе от одного элемента к другому в группах и подгруппах наблюдается не простое повторение свойств, а их закономерное изменение.
13.3. Электронные формулы элементов
Разберѐм и составим несколько электронных формул (см. Лекцию 1) и на их основе постараемся объяснить свойства элементов.
Пример 1.
Какому элементу принадлежит электронная формула
1s2
2s2
2p6
3s1
и каковы свойства данного элемента?
В атоме данного элемента 11 электронов. Атом нейтрален, следовательно, заряд его ядра равен +11. На внешнем уровне атома данного элемента (3s) находится 1 электрон. Это значит, что атом обладает малой энергией ионизации и является активным металлом.
Это щелочной металл натрий.
Пример 2
. Какая конфигурация соответствует элементу №17? Каковы свойства этого элемента? Какую электронную конфигурацию имеет анион данного элемента?
Заряд ядра элемента №17 равен +17. В нейтральном атоме этого элемента 17 электронов. Данный элемент находится в 3 периоде (так как в первом периоде два элемента, а во втором – восемь). Главное квантовое число для электронов данного атома принимает три значения: n = 1, 2,3. На первом уровне находится 2 s- электрона, на втором – 8 (2 s- и 6 р- электронов), а на третьем – 7 электронов (2 s- и 5 p-электронов). У данного элемента имеется также 3d-подуровень, так как для n=3 орбитальное квантовое число l имеет три значения: 0 (s-подуровень), 1 (p-подуровень) и 2 (d-подуровень). Следовательно, электронная формула элемента №17 имеет вид: 1s2
2s2
2p6
3s2
3p5
3d0
. Это р-элемент с ярко выраженными неметаллическими свойствами, так как на внешнем электронном уровне у него имеется семь электронов, причѐм один неспаренный. Это одновалентный элемент, но у него имеется 5 свободных d-орбиталей, на которые могут последовательно распариваться 2, 4 или шесть электронов. Следовательно, данный элемент может быть 1-, 3-, 5- и 7-валентным. Данный элемент легко присоединяет один электрон (которого ему не хватает до октета) и превращается в однозарядный анион. Электронная формула аниона данного элемента (хлора) имеет вид: 1s2
2s2
2p6
3s2
3p6
3d0
.
13.4. Краткий обзор свойств s-, p-, d- и f-элементов
Сочетание атомов одного элемента есть простое вещество
. Простые вещества могут быть металлами и неметаллами
. Металлы обладают наименьшими энергиями ионизации
и легко теряют свои электроны. Энергия ионизации неметаллов, напротив, высока; они обладают наибольшим сродством к электрону, то есть способны легко присоединять электроны других элементов. У элементов первой - тринадцатой групп Периодической системы Д.И. Менделеева преобладают металлические свойства. Для простых веществ этих элементов характерно металлическое состояние и такие физические свойства, как ковкость, электропроводность, металлический блеск и т.п. В сложных соединениях эти элементы выполняют катионную функцию, так как обладают минимальной энергии ионизации. Химические свойства: взаимодействие с водой, кислотами, щелочами, неметаллами.
В Периодической таблице границу между металлами и неметаллами можно условно провести по диагонали от бора к астату
: металлы будут располагаться слева внизу от этой диагонали, а неметаллы – справа вверху.
С увеличением порядкового номера элемент, как в периоде, так и в группе, свойства неметаллов ослабевают
, свойства металлов усиливаются
. Изменение химической активности объясняется изменением атомных радиусов, потенциалов ионизации атомов и другими характеристиками (такими, например, как теплота возгонки простых веществ и энергия кристаллической решетки). Самым активным металлом в Периодической системе является рубидий (цезий и франций - радиоактивные элементы со специфическими свойствами), а самым активным неметаллом – фтор.
Свойства s-элементов:
К s-элементам относятся щелочные и щелочноземельные металлы I и II групп Периодической системы. К элементам первой группы относятся водород, литий, натрий, калий, рубидий, цезий и франций
. Самыми распространѐнными щелочными металлами являются натрий и калий. Цезий и франций – радиоактивные элементы. Щелочные металлы первой группы в своих соединениях одновалентны; они являются очень активными металлами и бурно реагируют с водой с образованием щелочей и выделением водорода:
Na + H2
O → NaOH + H2
Реакция протекает с выделением большого количества энергии, а в результате бурного взаимодействия выделяющегося водорода с кислородом воздуха может произойти взрыв
. Ещѐ более бурно протекают реакции взаимодействия щелочных металлов с активными неметаллами, например, с водородом, хлором или кислородом c с образованием гидридов, пероксидов или солей:
2К+ Н2
→ 2КН; 2Li + O2
→ Li2
O2
; 2Na + Cl2
→ 2NaCl
Пероксиды щелочных металлов реагируют с водой образованием пероксида водорода и щелочей:
Na2
O2
+ H2
O → H2
O2
+ NaOH
При взаимодействии пероксида натрия с диоксидом углерода выделяется кислород:
Na2
O2
+ СO2
→ O2
+ Na2
СO3
Пероксиды крайне реакционноспособны и взрываются даже при простом соприкосновении с окислителями. Нормальные оксиды щелочных металлов Na2
O, Li2
O, К2
O и т.п. можно получить только косвенным путѐм. Гидроксиды щелочных металлов растворяются в воде и являются самыми сильными основаниями и часто называются едкими щелочами. Щѐлочи, в особенности гидроксид натрия (едкий натр, каустическая сода), производятся методом электролиза. Можно также получить едкий натр в обменной реакции, в которой малорастворимый карбонат натрия осаждается из раствора:
Na2
CO3
+ Ca(OH)2
→ 2NaOH + CaCO3
Щелочные металлы, особенно натрий и калий, образуют множество солей разных кислот. Их соли хорошо растворимы в воде и являются сильными электролитами. Водород, который тоже находится в первой группе, иногда называют газообразным металлом, так как многие его свойства сходны с металлическими. Так, например, он активно реагирует с неметаллами:
H2
+ Cl2
→ 2HCl;
С другой стороны, водород по многим химическим свойствам сходен с неметаллами XVII группы (галогенами), так как существует в виде двухатомной молекулы и вступает в реакцию с металлами: 2Na + Н2
→ 2NaH
Поэтому водород часто помещают как в группе I, так и в группе XVII. Щелочноземельные металлы второй группы, - бериллий, магний, кальций, стронций
и барий
(радий
является радиоактивным элементном), - менее активны, чем металлы первой группы. Они реагируют с водой только при высокой температуре (с водяным паром) по схеме:
Ca + 2H2
O(г)
→ Ca(OH)2
+ H2
Оксиды этих металлов (за исключением оксида бериллия) реагируют с водой с образованием сильных оснований:
СаО + Н2
О → Са(ОН)2
(гашение извести);
BаО + Н2
О → Bа(ОН)2
Оксиды щелочноземельных металлов реагируют с кислотами, то есть носят основной характер; оксид бериллия растворяется также и в сильных кислотах, то есть является амфотерным соединением.
Гидроксид бериллия Be(OH)2
также проявляет не только основные, но и кислотные свойства, реагируя с сильными основаниями с образованием бериллатов:
Be(OH)2
+ 2NaOH → Na2
BeO2
+ 2H2
O
Сульфаты и карбонаты щелочноземельных металлов мало растворимы в воде, однако их кислые соли (См. Лекцию 15) хорошо растворяются. Присутствие в воде солей магния и кальция делает воду жѐсткой, так как даже при незначительном содержании в воде ионов кальция и магния в моющих средствах (растворимых натриевых и калиевых солях жирных карбоновых кислот) образуются нерастворимые кальциевые и магниевые соли, оседающие в виде белого налѐта. Природные воды делаются жѐсткими при растворении карбонатов щелочноземельных металлов, которые содержатся в известняках, в присутствии избытка диоксида углерода:
CaCO3
+ H2
O + CO2
→ Ca(HCO3
)2
Свойства р-элементов
:
К р-элементам относятся элементы XIII - XVIII групп Периодической системы. По свойствам большая часть р-элементов относится к активным неметаллам. Наиболее активными неметаллами являются элементы XVII группы фтор, хлор, бром
и йод
(астат
является радиоактивным элементом), которые называются галогенами,
или галоидами
(солеобразующими) за их свойство реагировать с металлами с образованием солей:
Fe + Cl2
→ FeCl2
; Cu + I2
→ CuI2
Ярко выраженные неметаллические свойства галогенов объясняются тем, что до октета им не хватает всего одного электрона, вследствие чего эти элементы обладают высоким сродством к электрону. Все галогены реагируют с водородом с образованием соответствующих газообразных галогеноводородов HF, HCl, HBr и HI, растворы которых в воде являются сильными кислотами. Всем галогенам, кроме фтора, присуща переменная валентность, что объясняется распариванием электронов с уровня 3р на уровень 3d. Эти элементы могут быть I, II, III, V и VII-валентными. Вследствие этого многие соединения галогенов проявляют окислительно-восстановительные свойства (См. Лекцию 15). Известны кислородсодержащие кислоты галогенов для всех валентных состояний. Наиболее важным для практического применения является хлор. Его применяют для обеззараживания воды, отбеливания тканей и бумаги и в иных целях. В промышленности хлор получают методом электролиза раствора хлорида натрия, а в лабораторной практике – окислением соляной кислоты HCl соединениями марганца: MnO2
+ 4HCl → MnCl2
+ Cl2
+ 2H2
O;
2KMnO4
+ 16HCl → 2KCl + 2MnCl2
+ 5Cl2
+ 8H2
O
При пропускании хлора в горячий раствор щелочи образуется хлорат и хлорид калия:
3Cl2
+ 6KOH → KClO3
+ 5KCl + 3H2
O
В ряду кислородных кислот хлора HOCl (хлорноватистая) HClO2
(хлористая), HClO3
(хлорноватая) HClO4
(хлорная) кислотные свойства усиливаются, а окислительные свойства ослабевают. При переходе от XVII группы к XVI, XV, XIV и XIII группам неметаллические свойства рэлементов постепенно ослабевают. Кислород
, р-элемент XVI группы, является самым распространѐнным в природе. Все элементы, кроме инертных газов, реагируют с кислородом с образованием оксидов. В соединениях кислород всегда двухвалентен. В пероксидах
, например, в пероксиде водорода Н2
О2
или натрия Na2
O2
, атомы кислорода образуют «кислородные мостики» -О-О-
. Такая связь непрочна, поэтому пероксиды неустойчивы и часто разлагаются со взрывом. Остальные рэлементы XVI группы, сера, селен
и теллур
(полоний
является радиоактивным элементом) обладают переменной валентностью II, IV и VI. Серу, селен и теллур иногда называют «халькогенами» («рождающими медь») поскольку они обычно присутствуют в медных рудах. Оксиды этих элементов имеют кислотный характер. Например, при растворении оксидов серы в воде образуются кислоты:
SO2
+ H2
O → Н2
SO3
(сернистая кислота);
SO3
+ H2
O → H2
SO4
(
серная кислота)
Поэтому диоксид серы SO2
и триоксид серы SO3
называют соответственно сернистым и серным ангидридами
(«лишѐнными воды кислотами»). При реакции кислотных оксидов со щелочами образуются соли:
2NaOH + SO2
→ Na2
SO3
+ Н2
О
Сера образует и другие кислоты, сероводородную H2
S, серноватистую, или тиосерную, H2
S2
O3
, пиросерную H2
S2
O7
, надсерную H2
S2
O8
и другие. Эти кислоты, как и сернистая кислота, нестойки и не существуют в свободном виде, однако их соли (сульфиты, сульфиды, тиосульфаты, пиросульфаты, персульфаты) очень устойчивы и широко используются.
Металлические руды часто являются сульфидами металлов, например, ZnS, SnS, Bi2
S3
.
Азот
, самый распространѐнный в природе р-элемент XV группы c электронной формулой 1s2
2s2
2p3
обладает уникальными химическими свойствами. Для него характерны валентности I, II, III, IV и V.
Одновалентное состояние атома азота (например, в соединении N2
O) объясняется спиновой теорией валентности, выдвинутой Лондоном в 1928 году. В соответствии с этой теорией, при образовании химической связи спины валентных электронов пределах одного подуровня могут взаимно компенсироваться. Так, у азота компенсируются спины двух из трѐх 2р-электронов. Пятивалентное состояние можно объяснить участие в связи, помимо трѐх неспаренных 2р-электронов, «неподеленной» пары 2s-электронов. Возникновение двухвалентного и четырѐхвалентного состояния, например, в молекулах NO и NO2
, объясняют тем, что в этих молекулах фактически реализуется трѐхвалентное или пятивалентное состояние, но при этом один из валентных электронов азота остаѐтся неспаренным.
В воздухе содержится 75,6% молекулярного азота N2
. Реакция синтеза аммиака из азота воздуха с образованием аммиака по схеме:
N2
+ H2
→ NH3
называется реакцией связывания азота. При растворении аммиака в воде образуется гидроксид аммония, со свойствами слабого основания: NH3
+ H2
O → NH4
OH
Реагируя с кислотами, гидроксид аммония образует соли аммония:
2NH4
OH + H2
SO4
→ (NH4
)2
SO4
+ 2H2
O
Из кислородных соединений азота наибольшее применение находит азотная кислота HNO3
. При реакции еѐ с металлами образуются различные соединения азота:
3Cu + 8HNO3
(р) → 3Cu(NO3
)2
+ 2NO + 4H2
O
Продуктами реакции, в зависимости от активности металла и концентрации азотной кислоты, могут быть также диоксид азота NO2
, закись азота N2
O и аммиак NH3
. Соли азотной кислоты, нитраты, находят широкое применение. При нагревании нитраты разлагаются, причѐм продукты разложения зависят от активности входящего в состав соли металла. Соли металлов активнее магния разлагаются с образованием солей азотистой кислоты, нитритов, и кислорода; соли менее активных металлов (от магния до меди) образуют оксиды, а металлы, менее активные, чем медь, выделяются в свободном виде:
2NaNO3
→ 2NaNO2
+ O2
; 2Pb(NO3
)2
→ 2PbO + 4NO2
+ O2
2AgNO3
→ 2Ag + 2NO2
+ O2
Смесь концентрированной азотной и хлористоводородной кислот в отношении 1:3 называется «царской водкой»:
HNO3
+ 3HCl ↔ NOCl + Cl2
+ 2H2
O
Царская водка растворяет даже золото и платину; активным действующим началом этой смеси являются хлористый нитрозил NOCl и хлор в момент выделения. Со многими металлами азот образует нитриды
, которые чаще всего получают накаливанием металла в среде аммиака:
2Al + 2NH3
→ 2AlN + 3H2
Для фосфора, мышьяка, сурьмы
и висмута
характерны валентности III и V. Примеры соединений фосфора – фосфин PH3
, фосфорный ангидрид P2
O5
, ортофосфорная кислота H3
PO4
, отрофосфат натрия Na3
PO4
. При взаимодействии фосфорного ангидрида с водой можно получить метафосфорную кислоту HPO3
, пирофосфорную кислоту H4P2O7 или ортофосфорную кислоту H3
PO4
, которую часто называют просто фосфорной. Эту кислоту можно получить при взаимодействии фосфора с азотной кислотой в водном растворе:
3P + 5HNO3
+2H2
O → 3H3
PO4
+ 5NO
Соли азотной и фосфорной кислот используют в качестве удобрений, так как азот и фосфор, наряду с углеродом, водородом и кислородом – важные биогенные
элементы.
Неметаллические свойства элементов XV группы при переходе от фосфора к висмуту ослабевают, а металлические усиливаются. Так, мышьяк, сурьма и висмут в низшей валентности III образуют, подобно металлам, соли, в частности, хлориды AsCl3
, SbCl3
и BiCl3
.
Усиление металлических свойств при увеличении атомного номера еще нагляднее выражено у р-элементов XIV группы – углерода, кремния, германия, олова
и свинца
. Углерод в обычных условиях инертен, но при высоких температурах становится химически активным. У углерода две характерные валентности – II и IV, соответствующие оксиды – СО и СО2
. Диоксид углерода имеет кислотный характер и реагирует с водой с образованием угольной кислоты:
H2
O + CO2
↔ H2
CO3
Угольная кислота неустойчива, но еѐ соли, карбонаты, находят широкое применение. С металлами углерод вступает в реакцию только при высоких температурах с образованием карбидов
, например, CaC, W2
C. Из соединений углерода с неметаллами можно отметить сероуглерод CS2
, образующийся при пропускании паров серы через слой раскалѐнного угля, а также цианистый водород HCN, раствор которого в воде является нестойкой кислотой, соли которой, цианиды, напротив, устойчивы. Углерод является уникальным элементом, на основе которого образуются органические соединения (См. Лекцию 6), то есть является основным элементом для всей органической жизни. Ближайший аналог углерода, кремний, занимает третье место (после кислорода и водорода) по распространѐнности в природе. Основная часть земной коры состоит из силикатов
с общей формулой MeSiO3
. Силикаты являются солями кремниевой кислоты H2
SiO3
. Диоксид кремния SiO2
, или кремнезѐм, встречается в виде кварца, основной составной части обычного песка. При взаимодействии диоксида кремния с кремнием при высоком давлении можно получить также оксид кремния SiO, но это вещество неустойчиво, так как для кремния двухвалентное состояние нехарактерно. С металлами кремний образует силициды
(например, Mg2
Si), с галогенами – галиды SiCl4
, SiF4
, SiI4
SiBr4
.
Соединения кремния с водородом, силаны
, различны по составу (например, SiH4
, Si2
H6
… Si8
H18
) и являются аналогами углеводородов ряда метана, однако отличаются от них высокой реакционной способностью, в частности, воспламеняются на воздухе и сгорают с образованием диоксида кремния и воды. Гидриды кремния состава Sin
H2n
и Sin
Hn
можно считать аналогами непредельных углеводородов, так как они также способны к полимеризации
(см. Лекцию 6) . Полисилен (SiH2
)n можно назвать неорганическим полимером
. Олово и свинец по физическим свойствам являются типичными металлами, тогда как германий сходен с кремнием и является. Для всех трѐх элементов характерны валентности II и IV, они образуют соответствующие оксиды, соли и гидроксиды.
Из р-элементов XIII группы только бору
присущи ярко выраженные неметаллические свойства. В своих соединениях он, как правило, трѐхвалентен. Наиболее распространѐнными его соединениями являются ортоборная кислота H3
BO3
и соль тетраборной кислоты бура Na2
B2
O7
∙10H2
O. В обычных условиях бор инертен, но при высоких температурах соединяется с металлами, образуя бориды
(например, MgB2
), или с неметаллами с образованием оксида B2
O3
и галидов (например, BCl3
). При воздействии кислот на сплавы бора с магнием образуются соединения бора с водородом, или бораны
. Горение диборана B2
H6
, сопровождается выделением огромного количества теплоты, поэтому его можно использовать в качестве ракетного топлива. Алюминий
по распространѐнности стоит на четвѐртом месте (после кислорода, водорода и кремния), это металл, обладающий ценными свойствами, в частности прочностью и лѐгкостью. Его получают методом электролиза расплава глинозѐма Al2
O3
. Плѐнка этого оксида на поверхности металла делает алюминий устойчивым к действию кислот, хотя сильные щелочи легко растворяют алюминий с образованием алюминатов и водорода:
2Al + 2NaOH + 2H2
O → 2NaAlO2
+ 3H2
Гидроксид алюминия реагирует как с кислотами с образованием солей алюминия, так и со щелочами с образованием алюминатов. Галлий, индий
и таллий
реагируют с неметаллами активнее, чем алюминий, и хорошо растворяются в кислотах. У таллия имеются соединения, в которых он одновалентен. Кислотный характер гидроксидов галлия, индия и таллия выражен гораздо слабее, чем у алюминия.
Р-элементы XVIII группы, - гелий, неон, аргон, криптон
, ксенон
и радиоактивный радон
, - относятся к инертным газам;
они практически не вступают в химические реакции, потому что в их атомах все электроны спарены. Эти одноатомные газы долгое время считались химически абсолютно инертными, однако в 1962 году было обнаружено, что в определѐнных условиях они способны соединяться с фтором с образованием фторидов различного состава. Например, для ксенона, образующего самые устойчивые соединения, известны фториды XeF2
XeF4
и XeF6
. Был также получен триоксид ксенона XeO3
.
Свойства d-элементов:
В группах с III по XII располагаются d-элементы
. По физическим свойствам это типичные металлы; в стандартных условиях все они, кроме ртути, находятся в твѐрдом состоянии. Химические свойства переходных элементов определяются строением внешнего электронного уровня dm
sn
, где m (количество электронов на d-подуровне) изменяется от 1 до 10, а n (количество электронов на s-подуровне) - от 2 до 0, так как у многих переходных элементов имеет место «проскок» s-электрона на d-подуровень. Электроны внешнего уровня у всех d-элементов находятся достаточно далеко от ядра, поэтому энергия ионизации как s-, так и d-электронов невелика. Поэтому в образовании связи могут участвовать не только s-, но и р-электроны. Это значит, что многие переходные р-элементы обладают переменной валентностью, которая может варьироваться от I до VIII. Проявляя низкие валентности (I-III), dэлементы ведут себя как металлы; проявляя высшие валентности (IVVIII), они обладают свойствами неметаллов. Именно поэтому их часто называют переходными элементами
. Это название условно, поскольку двойственными свойствами обладают также многие pэлементы. Вот почему в «короткой» Периодической системе, где элементы подразделяются на 8 групп, d-элементы помещают в «типические» подгруппы, сравнивая их свойства со свойствами pэлементов, хотя электронное строение у них совершенно разное. По химическим свойствам переходные металлы сильно отличаются друг от друга. Так, металлы платиновой группы (рутений, родий, палладий, осмий иридий
и платина
), отличаются высокой химической устойчивостью и не подвергаются коррозии; все они, кроме платины и палладия нерастворимы даже в царской водке. Перевести платиновые металлы в растворимое состояние можно только сплавлением их со щелочами в присутствии окислителей. Палладий в соединениях, например, в хлориде палладия PdCl2
, проявляет валентность II; для родия и иридия характерна валентность III, их гидроксиды Ph(OH)3
и Ir(OH)3
являются слабыми основаниями. Четырѐхвалентное состояние характерно для платины, например, при растворении платины в насыщенной хлором соляной кислоте можно получить платинохлористоводородную кислоту H2
PtCl6
. Осмий и рутений чаще всего проявляют валентность VI (K2
OsO4
, K2
RuO4
) и VIII (RuO4
, OsO4
). К
платиновым металлам примыкают «благородные» металлы золото
и серебро
. Они не растворяются в кислотах, которые не обладают окислительными свойствами, и не окисляются на воздухе. В кислотахокислителях серебро растворяется легко, тогда как золото можно растворить только в царской водке или в насыщенной хлором соляной кислоте. В своих соединениях серебро одновалентно (AgNO3
, AgCl) , а золото проявляет как валентность I (AuCl, AuBr), так и валентность III (AuCl3
, AuBr3
). Медь
на воздухе покрывается плѐнкой углекислых солей, при нагревании реагирует с кислородом с образованием оксидов, растворяется в азотной кислоте. Для меди характерны две валентности – I (например, оксид одновалентной меди Cu2
O) и II (CuO). Самое широкое применение находят соединения двухвалентной меди; гидроксид меди (II) Cu(OH)2
нерастворим и обладает свойствами слабого основания, из солей меди (II) наибольшее практическое значение имеет пятиводный сульфат меди, или медный купорос CuSO4
∙5H2
O. Из триады d-элементов в составе железа, никеля
и кобальта
исключительное практическое применение имеет железо. Его мировое производство превышает 500 миллионов тонн. По химическим свойствам железо, никель и кобальт относятся к металлам средней активности, химически чистые металлы устойчивы к коррозии, хотя содержащее примеси железо подвергается атмосферной коррозии под воздействием влаги, кислорода воздуха и углекислоты (см. Лекцию 4). При высоких температурах активность этих металлов возрастает, например, железо разлагает воду по схеме:
Fe(порошок) + H2O (г) → FeO + H2
В своих соединениях все металлы проявляют валентности II и III, хотя никель встречается в трѐхвалентном состоянии крайне редко. При растворении железа в кислотах образуются соли железа (II) например, FeCl2
, FeSO4
, Fe(NO3
)2
, которые могут легко окисляться с образованием солей трѐхвалентного железа: FeCl3
, Fe2
(SO4
)3
, Fe(NO3
)3
. Гидроксиды двухвалентных железа, кобальта и никеля нерастворимы и являются слабыми основаниями; их можно получить взаимодействием соответствующих солей со щелочами:
NiCl2
+ 2NaOH → Ni(OH)2
+ 2NaCl
Гидроксид двухвалентного железа легко окисляется кислородом воздуха с образованием гидроксида трѐхвалентного железа:
4Fe(OH)2
+ O2
+ 2H2
O → 4Fe(OH)3
Гидроксид кобальта Co(OH)2
окисляется гораздо медленнее, а гидроксид никеля Ni(OH)2
не реагирует с кислородом.
Около 90% всего добываемого марганца
потребляется для производства легированных сталей. Сплавы железа с марганцем необычайно прочны и используются для производства деталей, от которых требуется высокое сопротивление ударам. Марганец - активный металл, реагирующий с галоидами, серой, азотом, фосфором и другими неметаллами. Он растворяется в кислотах с образованием солей, например, MnCl2
. Кроме двухвалентного состояния, для марганца характерны валентности III, IV, VI и VII. В каждом состоянии он образует соответствующий оксид: оксид марганца (II) MnO, оксид марганца (III) Mn2
O3
, диоксид марганца MnO2
, марганцовистый ангидрид MnO3
и марганцовый ангидрид MnO4
. В ряду соединений типа Mn-O-H: Mn(OH)2
, Mn(OH)3
, Mn(OH)4
, H2
MnO4
(марганцовистая кислота) и HMnO4
(марганцовая кислота) слева направо усиливаются кислотные свойства, а слева направо – основные. Соли марганцовистой кислоты, или манганаты
, неустойчивы и распадаются с образованием диоксида марганца и соли марганцовой кислоты по схеме:
3K2
MnO4
+ 2H2
O → MnO2
+ 2KMnO4
+ 4KOH
Наиболее важным соединением марганца (VII) является перманганат калия
KMnO4
, который используется в качестве сильного окислителя (см. Лекцию 14). Широкое применение находят также соли марганца (II). При действии на такие соли щелочей осаждается белый гидроксид марганца (II), который на воздухе буреет с образованием гидроксида марганца (IV):
MnCl2
+ 2KOH → Mn(OH)2
+ 2KCl; 2Mn(OH)2
+ O2
+ 2H2
O → 2Mn(OH)4
Для аналогов марганца технеция
и рения
наиболее характерно семивалентное состояние.
Хром, молибден
и вольфрам
являются довольно распространѐнными элементами и находят широкое применение в качестве легирующих элементов при производстве специальных сталей. Соединения хрома используются также для хромирования
металлических изделий для повышения их сопротивления износу. Все три металла очень устойчивы к коррозии и тугоплавки. Получают их методом алюмотермии
при взаимодействии соответствующих оксидов с порошком алюминия: Cr2
O3
+ 2Al →Al2
O3
+ 2Cr
Реакция протекает с выделением теплоты, что способствует восстановлению более ценного металла. Для хрома характерны валентности III и VI, а для вольфрама и молибдена – валентность VI, хотя существуют соединения, в которых эти элементы проявляются также валентности II, IV и V. Проявляя валентность VI, хром и его аналоги ведут себя как неметаллы и образуют кислоты типа H2
ЭО4
и Н2
Э2
О7
. Эти кислоты нестойки, однако их соли находят широкое применение, в особенности хромат калия K2
CrO4
, дихромат калия K2
Cr2
O7
а также вольфрамат и молибдат натрия Na2
WO4
и Na2
MoO4
.
Дихроматы переходят в хроматы, и наоборот, в результате реакций:
K2
Cr2
O7
+ 2KOH → 2K2
CrO4
+ H2
O; 2K2
CrO4
+ H2
SO4
→ K2
Cr2
O7
+ K2
SO4
+ H2
O
При осторожном растворении дихроматов в концентрированной серной кислоте получают хромовую смесь, эффективное средство для мытья лабораторной химической посуды. Хромовая смесь растворяет любые загрязнения вследствие наличия в ней хромового ангидрида CrO3
: K
2Cr
2O
7 + H
2SO
4(конц.) → 2CrO
3 + K
2SO
4 + H
2O
Из соединений трѐхвалентного хрома наиболее важны соли, например, хлорид CrCl3
и сульфат Cr2
(SO4
)3
. Из раствора соли хрома (III) можно осадить малорастворимый гидроксид хрома (III), который имеет амфотерный характер, так как реагирует с кислотами с образованием солей хрома (III), а при растворении в щелочи образует соли хромистой кислоты, или хромиты:
Cr(OH)3
+ 3HCl → CrCl3
+ 3H2
O; Cr(OH)3
+ KOH → KCrO2
+ 2 H2
O
К наиболее распространѐнным соединениям двухвалентного хрома относится оксид CrO и фторид CrF2
. Для вольфрама и молибдена наиболее характерным является шестивалентное состояние.
Для ванадия, ниобия
и тантала
наиболее характерна валентность V, хотя известны соединения, в которых эти металлы проявляют валентности II, III и IV. В чистом виде эти металлы химически устойчивы, не подвергаются коррозии. Основной областью их применения является металлургия специальных сталей. При реакции ангидридов ванадия, ниобия и тантала со щелочами получают соответствующие ванадаты, ниобаты и танталаты:
V2
O5
+ 2NaOH → 2NaVO3
+ H2O; Nb2
O5
+ NaOH → NaNbO3
+ H2O; Ta2
O5
+ NaOH → NaTaO3
+ H2O
Оксид ванадия (V) имеет ярко выраженный кислотный характер, образуя при растворении в воде ванадиевую кислоту; оксиды и гидроксиды ниобия и тантала амфотерны.
Титан, цирконий
и гафний
обычно получают путѐм восстановления их хлоридов магнием по схеме:
TiCl4
+ 2Mg → 2MgCl2
+ Ti
По физическим свойствам эти элементы являются типичными металлами, в обычных условиях обладают высокой химической стойкостью, но при высоких температурах химически очень активны и соединяются не только с галогенами, кислородом и серой, но и с углеродом и азотом. Титан и цирконий используются в качестве самостоятельных конструкционных материалов в самолѐтостроении (титан) и в производстве атомных реакторов (цирконий). Гафний используется в качестве катода в телевизионных кинескопах и в ядерной энергетике как поглотитель нейтронов. В своих важнейших соединениях эти металлы обычно четырѐхвалентны. Гидроксид титана Ti(OH)4
имеет амфотерный характер, хотя его основные, а особенно кислотные свойства выражены слабо. При переходе к гидроксидам циркония и гафния кислотные свойства ещѐ более ослабевают, а основные усиливаются. Из других соединений наибольшее значение имеют галиды TiCl4
, ZrCl4
и HfCl4
. Соединения, в которых эти элементы проявляют валентности II и III немногочисленны и неустойчивы. Цинк, кадмий
и ртуть
присутствуют в природе преимущественно в виде сульфидов, поэтому при получении этих металлов сначала превращают сульфиды в оксиды, а затем восстанавливают оксиды углем:
2ZnS + 3O2
→ 3SO2
+ 2 ZnO; ZnO + C → Zn + CO
Оксид ртути HgO при высоких температурах разлагается, поэтому получение ртути сводится к одной реакции:
HgS + O2
→ SO2
+ Hg
Для цинка и кадмия характерна валентность II, а для ртути – валентности I и II. Соединения кадмия и ртути очень ядовиты. Поверхность металлов быстро покрывается защитной оксидной плѐнкой; в разбавленных соляной и серной кислотах цинк растворяется легко, кадмий – медленно, а ртуть нерастворима. В азотной кислоте все металлы растворяются хорошо. Например, ртуть реагирует по схеме: 3Hg + 8HNO3
→ 3Hg(NO3
)2
+ 2NO + 4 H2
O
В отличие от кадмия и ртути, цинк растворяется в концентрированных растворах щелочей с образованием цинкатов
:
Zn + 2 NaOH → Na2
ZnO3
+ H2
В отличие от цинка и кадмия, у ртути имеются соединения, в которых она формально одновалентна (Hg2
Cl2
, Hg2
S); на самом же деле в таких соединениях содержится «ртутный мостик» -Hg-Hg-. Соединения, имеющие в своѐм составе такую группировку, менее ядовиты, чем соединения ртути (II). Например, HgCl2
– сулема, Hg2
Cl2
– каломель.
Многие металлы растворяются в ртути с образованием амальгам
, которые легко размягчаются при нагревании. Свойства содержащегося в амальгаме металла практически не изменяются. Например, при соприкосновении амальгамы натрия с водой, как и случае металлического натрия, образуется щѐлочь и выделяется водород, хотя реакция идет значительно спокойнее. На этом основании применение амальгамы натрия в качестве восстановителя.
Скандий, иттрий
и лютеций
в своих соединениях трѐхвалентны. Оксиды этих металлов легко соединяются с водой, образуя гидроксиды Sc(OH)3
, Y(OH)3
Lu(OH)3
, которые являются слабыми основаниями.
Все d-элементы образуют комплексные соединения
(см. Лекцию15), так как для них характерна донорно-акцепторная связь (см. Лекцию15).
Многие d-элементы имеют также каталитические
свойства.
Свойства f-элементов:
К f-элементам относятся семейства лантанидов в шестом периоде и актинидов в седьмом периоде. Их обычно выносят за пределы собственно таблицы в виде отдельных строк. Лантан
и актиний
занимают в этом ряду особое место, так как по строению наружных электронных оболочек (5d1
6s2
у лантана и 6d1
7s2
у актиния) относятся к d-элементам. Дело в том, что элемента, содержащего в своѐм атоме один f-электрон, не существует в природе. Этот f-электрон «проскакивает» на подуровень 5d у лантана и 6d у актиния. Лютеций и лоуренсий имеют такое же строение внешних электронных слоѐв, как лантан и актиний (5d1
6s2
у лютеция и 6d1
7s2
у лоуренсия), однако, в отличие от лантана и актиния, у лютеция и лоуренсия полностью заполнены электронами подуровни 4f и 5f. Все f-элементы шестого периода по химическим свойствам являются активными металлами
и похожи друг на друга. Это объясняется тем, что они имеют одинаковое строение внешних d- и s- подуровней, а разнятся только строением глубинного f-подуровня. Эти глубинные подуровни хорошо экранируются внешними электронами, так что изменение числа f-электронов почти не влияет на химические свойства атомов.
Лантан и церий, особенно в измельчѐнном виде, мгновенно окисляются на воздухе, поэтому в некоторых зажигалках используется кремень, содержащий эти металлы. Лантаниды реагируют с водой с образованием водорода и малорастворимых гидроксидов основного характера:
2La + 6H2
O → 2La(OH)3
+ 3H2
; La(OH)3
+ HCl → LaCl3
+ 3 H2
O
Для лантанидов в соединениях характерна валентность III, хотя церий, празеодим и тербий проявляют также валентность IV, а самарий, европий и иттербий – валентность II. С возрастанием атомного номера атомные радиуса лантанидов уменьшаются; это явление называется лантанидным сжатием
и объясняется ростом заряда ядра. Лантаниды относятся к редкоземельным элементам, они встречаются в природе в очень рассеянном виде и всегда смешаны друг с другом и с иттрием. В настоящее время лантан, церий, неодим, празеодим, самарий, европий, тербий, диспрозий и сопутствующий лантанидам редкоземельный dэлемент иттрий находят широкое применение в качестве материала для изготовления жидкокристаллических дисплеев, а также защитных экранов для поглощения излучений атомных реакторов.
Актиний и актиниды
являются радиоактивными элементами, так как претерпевают радиоактивный распад. Фактически, все элементы седьмого периода радиоактивны. Радиоактивность сама по себе не влияет на химические свойства, однако радиоактивные элементы представляют интерес не как химические вещества, а в качестве ядерного топлива, так как при распаде ядер выделяется колоссальная энергия. В природе встречаются только четыре элемента седьмого периода – актиний, торий, протактиний и уран. Химические свойства этих элементов изучены довольно хорошо, тогда как свойства остальных элементов искусственного происхождения затрудняется малым временем их жизни и невозможностью получить достаточное для химических исследований количество вещества. Актиниды гораздо больше отличаются друг от друга по своим валентным состояниям, нежели лантаниды; валентность актинидов варьируется от II до VII. Так, для урана наиболее характерна валентность VI; уран в виде химического соединения обычно продаѐтся в виде азотнокислого уранила UO2
(NO3
)2
.
Периодический закон - фундамент химии
, в первую очередь химии неорганической. Он помогает решению множества задач: это и понимание химических свойств простых веществ и их соединений, и синтез веществ с заданными свойствами, и подбор оптимальных условий для проведения различных химических процессов, и многое другое.
13.5. Вопросы и задания:
1. Сформулируйте периодический закон Д.И. Менделеева в оригинальной и современной трактовке.
2. Объясните происхождение «магических чисел» 2, 8, 18 и 32 в системе Менделеева. Каким было бы следующее «магическое число»?
3. Какие элементы относятся s-, р-, d- и f- элементам?
4. Напишите электронные формулы атомов элементов с порядковыми номерами 9, 15, 18, 27. К какому электронному семейству относится каждый из этих атомов?
5. Атомам каких элементов соответствуют следующие электронные формулы? 1s2
2s2
; 1s2
2s2
2p5
; 1s2
2s2
2p6
3s1
; 1s2
2s2
2p6
3s2
3p6
3d3
4s2
; 1s22s22p63s23p63d104s1; 1s22s22p63s23p63d104s24p64d105s25p65d16s2;
1s22s22p63s23p63d104s24p64d104f145s25p65d16s2
6. Приведите примеры 4-х частиц (атомов и ионов) с электронной конфигурацией 1s2
2s2
2p6
.
7. Как изменяются свойства элементов в периоде и группе с увеличением порядкового номера?
8. Определите пропущенные вещества в следующей реакции:
 Na
2
O
2
... H
2
O
2
... Na
2
O
2
... H
2
O
2
...
9. Определите вещества Х1
и Х2
в цепочке превращений:
 Ca X
1 CaCO
3 CO
2 H
2O
X
2 . Ca X
1 CaCO
3 CO
2 H
2O
X
2 .
10. Каким методом получают хлор в лабораторных условиях?
11.  Какие вещества пропущены в схеме химической реакции Cl
2
... KClO
3
... H
2
O
? Какие вещества пропущены в схеме химической реакции Cl
2
... KClO
3
... H
2
O
?
12. Определите вещества X1
, X2
и X3
в цепочках превращений:
 а) Fe
HCl
X
1 NaOH
X
2 t
0 X
3; б) FeSO
4 KMnO
4 X
1 KOH
X
2 t
o
X
3; а) Fe
HCl
X
1 NaOH
X
2 t
0 X
3; б) FeSO
4 KMnO
4 X
1 KOH
X
2 t
o
X
3;
в)Fe
(OH
)2
 3 3
13. Какие продукты реакции образуются при термическом разложении нитрата калия KNO3
и нитрата меди Cu(NO3
)2
?
14. Какие вещества образуются при окислении фосфора азотной кислотой?
15. Гидроксид какого элемента в следующем ряду: бериллий, магний, бор, литий, обладает амфотерными свойствами (реагирует как с кислотами, так и со щелочами)?
16. Укажите, какой оксид растворяется в воде с образованием кислоты:
P2
O5
, K2
O, N2
O, SiO2
.
17. Что является конечным веществом X3
в цепочке превращений
Zn
 X
3
? X
3
?
18. Определите вещество X в цепочке превращений: MnCl
2
 Mn
(OH
)4
. Mn
(OH
)4
.
19. Определите, для какого металла (меди, хрома, железа или цинка) характерны реакции, протекающие по схемам:
а) Na2
ЭO4
+H2
SO4
→ Na2
Э2
O7
+Na2
SO4
+H2
O;
б) Na2
Э2
O7
+ H2
SO4
(к) → 2ЭO3
+ Na2
SO4
+ H2
O
20. Определите, в каком процессе образуется щелочь: а) при горении магния в воде; б) при разложении воды раскалѐнным железом; в) при растворении хлористого водорода в воде; при растворении негашѐной извести в воде.
Лекция 14. Кислотные, основные и окислительновосстановительные свойства веществ
14.1. Кислоты и основания
Кислотами
называются соединения, способные в растворах отщеплять ионы водорода. Кислоты делятся на бескислородные (HCl, HF, H2
S, HCN и т.п.) и кислородсодержащие (H2
SO4
, H3
PO4
, HNO3
и пр.) В зависимости от числа входящих в молекулу кислоты атомов водорода кислоты подразделяют на одноосновные
, двухосновные и трехосновные
. В нижеследующей Таблице 14.1 приведены названия некоторых наиболее распространенных кислот и их солей:
Таблица 14.1. Некоторые распространенные кислоты и их соли
| Название кислоты
|
Формула кислоты
|
Названия нормальных (средних)
солей
|
| Азотная
|
HNO3
|
Нитраты
|
| Азотистая
|
HNO2
|
Нитриты
|
| Борная (ортоборная)
|
H3
BO3
|
Бораты
|
| Бромистоводородная
|
HBr
|
Бромиды
|
| Двухромовая
|
H2
CrO4
|
Дихроматы
|
| Йодистоводородная
|
HJ
|
Йодиды
|
| Йодноватая
|
HJO3
|
Йодаты
|
| Кремниевая
|
H2
SiO3
|
Силикаты
|
| Марганцовая
|
HMnO4
|
Перманганаты
|
| Метафосфорная
|
HPO3
|
Метафосфаты
|
| Мышьяковая
|
H3
AsO4
|
Арсенаты
|
| Мышьяковистая
|
H3
AsO3
|
Арсениты
|
| Ортофосфорная
|
H3
PO4
|
Ортофосфаты
|
| Пирофосфорная
|
H4P2O7
|
Пирофосфаты
|
| Родановодородная
(тиоциановая)
|
HSCN
|
Роданиды (тиоцианаты)
|
| Серная
|
H2
SO4
|
Сульфаты
|
| Сернистая
|
H2
SO3
|
Сульфиты
|
| Сероводородная
|
H2
S
|
Сульфиды
|
| Тиосерная
|
H2S2O3
|
Тиосульфаты
|
| Пиросерная
|
H2S2O7
|
Пиросульфаты
|
| Надсерная
|
H2S2O8
|
Персульфаты
|
| Угольная
|
H2
CO3
|
Карбонаты
|
| Уксусная
|
CH3
COOH
|
Ацетаты
|
| Фтороводородная
(плавиковая)
|
HF
|
Фториды
|
| Хлоровододородная,
(соляная)
|
HCl
|
Хлориды
|
| Хлорная
|
HClO4
|
Перхлораты
|
| Хлорноватая
|
HClO3
|
Хлораты
|
| Хлорноватистая
|
HClO
|
Гипохлориты
|
| Хромовая
|
H2
CrO4
|
Хроматы
|
| Циановодородная
(синильная)
|
HCN
|
Цианиды
|
| Щавелевая
|
H2C2O4
|
Оксалаты
|
По исторически сложившейся традиции некоторые кислоты, также как и соли некоторых кислот, имеют тривиальные названия, отличающиеся от систематических.
Гидроксидами
называются вещества, состоящие из гидроксильной группы ОН и металла. Основные
гидроксиды (например, КОН) проявляют свойства оснований (отщепляя в водном растворе гидроксильные группы), кислотные гидроксиды (например, HONO2
) в водном растворе ведут себя как кислоты, отщепляя катион водорода. Амфотерные основания (например, Zn(OH)2
) проявляют как те, так и другие свойства. Гидроксиды, образованные металлами первой группы (главной подгруппы) LiOH, NaOH, KOH, RbOH хорошо растворимы в воде, очень активны и носят тривиальное название щелочей
. Растворимы и некоторые гидроксиды металлов второй группы - кальция и бария. В зависимости от количества гидроксильных групп, входящих в состав гидроксидов, их делят на однокислотные
(NaOH), двукислотные
(Zn(OH)2
) и т.д.
В свете теории электролитической диссоциации кислотами называются электролиты, диссоциирующие в растворах с образованием ионов водорода.
Для кислот вследствие наличия в их растворах ионов водорода характерны такие общие свойства, как:
Способность взаимодействовать с основаниями с образованием солей;
 Способность взаимодействовать с некоторыми металлами с выделением водорода; Способность взаимодействовать с некоторыми металлами с выделением водорода;
Способность изменять цвета индикаторов (в частности, вызывать красную окраску лакмуса); Кислый вкус.
В точки зрения теории электролитической диссоциации основания – это электролиты, диссоциирующие в растворах с образованием гидроксид – ионов. Вследствие этого основания имеют следующие общие свойства:
Способность взаимодействовать с кислотами с образованием солей;
Способность изменять цвета индикаторов (иначе, чем кислоты:
лакмус синеет);
Растворы оснований «мыльные» на ощупь.
Некоторые гидроксиды способны реагировать не только с кислотами, но и с основаниями. Например, при взаимодействии гидроксида цинка с соляной кислотой получается хлорид цинка:
Zn(OH)2
+ 2HCl = ZnCl2
+ 2H2
O;
а при его реакции с гидроксидом натрия образуется цинкат натрия: Zn(OH)2
+ 2NaOH = Na2
ZnO2
+ 2H2
O;
Такие гидроксиды (цинка, алюминия, свинца, олова и других металлов) называются амфотерными электролитами
. Явление амфотерности объясняется тем, что диссоциация таких гидроксидов возможна по как по основному, так и по кислотному механизму. Например, гидроксид цинка Zn(OH)2
можно представить в виде Н2
ZnO2
.
В общем случае в соединении М – О – Н основные свойства выражены тем сильнее, чем более активным металлом является атом М. И наоборот, чем сильнее атом М притягивает электроны, то есть проявляет неметаллические свойства, тем более усиливаются кислотные свойства вещества.
Катион водорода, лишѐнный единственного электрона, можно рассматривать как «голый протон». В соответствии с протонной теорией кислот и оснований
Бернстеда-Льюиса, кислотой является донор протона, а основанием – акцептор протона, то есть существует следующее соотношение:
Основание + Протон ↔ Кислота;
Связанные этим соотношением основание и кислота называются сопряженными
. Например, в реакции образования слабой уксусной кислоты:
СН3
СОО-
+ Н+
↔ СН3
СООН
сопряжѐнным с уксусной кислотой основанием можно считать ацетатанион СН3
СОО-
, а в реакции образования гидроксида аммония (слабого основания):
NH4
+
+ OH-
↔ NH4
OH
катион аммония является сопряжѐнным с этим основанием кислотой. В зависимости от партнера одно и то же вещество можно рассматривать или как кислоту, или как основание. Например, вода по отношению к аммиаку является кислотой, а по отношению к фториду водорода – основанием:
H2
O + NH3
↔ OH-
+ NH4
+
; HF + H2O ↔ H3O+ [18]
+ F-
При реакции уксусной кислоты с аммиаком в водном растворе образуется соль – ацетат аммония, которая диссоциирует на ацетатанион и катион аммония:
CH3
COOH + NH3
↔ CH3
COO-
+ NH4
+
В результате данной реакции происходит перенос протона от протонодонора - уксусной кислоты к протоноакцептору - молекуле аммиака. Однако среди продуктов реакции также имеется протонодонор – катион аммония, который в обратной реакции отдаѐт катион водорода, и протоноакцептор – ацетат-анион, который в обратной реакции принимает протон. Кислота и основание реагируют с переносом протона
, образуя другие кислоту и основание:
Кислота1
+ Основание1
↔ Кислота2
+ Основание2
В данном примере парами сопряженных кислот и оснований являются уксусная кислота и ацетат-ион, а также катион аммония и аммиак.
Сравнить кислоты по силе можно на основании констант их диссоциации
(см. Лекцию 2).
14.2. Водородный показатель
Многие реакции протекают только кислой или щелочной среде, то есть в присутствии в растворе катионов водорода [Н+
] или гидроксиланионов [ОН-
]; часто ход реакции и состав еѐ продуктов зависит от того, какой из этих ионов присутствует в растворе. Кислотность или щелочность среды определяется концентрацией ионов водорода или гидроксила в растворе в моль/л. В нейтральном растворе или в чистой воде концентрация присутствуют как катионы водорода, так и анионы гидроксила. Это объясняется диссоциацией молекул воды:
H2
O ↔ H+
+ OH-
(14.1)
Равновесие данного процесса сильно смещено влево, однако по электропроводности чистой воды можно вычислить концентрации катиона водорода и гидроксил-аниона в чистой воде, которые, как следует из (15.1), равны друг другу. При 250
С [H+
] = [OH-
] = 10-7
моль/л.
Константа диссоциации воды определяется соотношением:
К
дисс
 , (14.2) , (14.2)
 или H OH K
дисс
H
2
O
. или H OH K
дисс
H
2
O
.
Поскольку диссоциация воды очень мала, то концентрация недиссоциированных молекул воды в воде равна практически общей концентрации воды, то есть 55,55 моль/л (1000г: 18,02г/моль). Это величина постоянная и в разбавленных растворах. Заменяем правую часть уравнения новой константой КН2О
и получаем:
K
H
2O
 (14.3) (14.3)
Для воды и разбавленных водных растворов при постоянной температуре произведение концентраций ионов водорода и гидроксила есть величина постоянная. Эта величина называется ионным произведением воды.
Подставляя значения концентраций ионов при 250
С в (15.3), получаем для указанной температуры:
 K
H
2O
10 7
10 7
10 14
(14.4) K
H
2O
10 7
10 7
10 14
(14.4)
Ионное произведение воды позволяет определять кислотность или щелочность растворов посредством измерения концентрации только одного из ионов – либо катиона водорода, либо гидроксил-аниона. Для измерения кислотности и щелочности раствора была выбрана концентрация катионов водорода. Через эту концентрацию можно выразить и концентрацию гидроксила в том же растворе. Например, если в растворе содержится 0,01 моль/л щелочи (концентрация гидроксил-анионов равна 10-2
моль/л), то по ионному произведению воды можно найти концентрацию ионов водорода в этом же растворе:
 14 ; 14 ;
Растворы, в которых концентрации ионов водорода и гидроксила одинаковы и равны 10-7
моль/л называются нейтральными растворами
. В кислых растворах больше ионов водорода, а в щелочных – больше ионов гидроксила. Это значит, что в кислом
растворе концентрация ионов водорода должна быть больше 10-7
моль/л
, а в щелочном
– меньше 10-7
моль/л
, так как произведение концентраций ионов водорода и гидроксила есть величина постоянная в любом растворе (10-14
).
Более удобно выражать концентрацию катионов водорода в логарифмической форме
. Вместо концентрации указывают ее десятичный логарифм с обратным знаком. Эта величина называется водородным показателем
и обозначается рН
(пэ аш).
рН
 (14.5) (14.5)
Следовательно, в нейтральном растворе рН = 7; в кислом растворе рН меньше 7, а в щелочном растворе рН больше 7
.
Пусть, например, требуется определить рН в растворе, содержащем в 500 мл 1,825 г соляной кислоты HCl. Это значит, что в 1л раствора содержится 3,65 г, или 0,1 моль кислоты. Соляная кислота, как сильный электролит, диссоциирует практически нацело по схеме:
HCl → H+
+ Cl-
,
то есть в данном растворе концентрация ионов водорода равна концентрации кислоты, или 0,1М. Находим рН раствора: рН = -lg10-1
= 1.
  Для измерения кислотности и щѐлочности растворов можно было выбрать не водородный, а гидроксильный показатель рОН, то есть отрицательный логарифм концентрации гидроксил-анионов в растворе. Водородный и гидроксильный показатель связаны соотношением: pH pOH
14. В нейтральном растворе pH pOH
7, в кислом растворе pH
< pOH
, а в щелочном pH
> pOH
. Для измерения кислотности и щѐлочности растворов можно было выбрать не водородный, а гидроксильный показатель рОН, то есть отрицательный логарифм концентрации гидроксил-анионов в растворе. Водородный и гидроксильный показатель связаны соотношением: pH pOH
14. В нейтральном растворе pH pOH
7, в кислом растворе pH
< pOH
, а в щелочном pH
> pOH
.
Для измерения реакции раствора (рН) существуют различные способы. Приближено реакцию среды можно определить при помощи специальных реактивов - индикаторов, - окраска которых меняется в зависимости от концентрации водородных ионов (рН). Наиболее распространенные индикаторы – это метиловый оранжевый (в кислой среде имеет красную окраску; в нейтральной оранжевую; в щелочной желтую), метиловый красный (красный – оранжевый - желтый), фенолфталеин (бесцветный – розовый – малиновый) и лакмус (красный – фиолетовый – синий). Часто используют смесь индикаторов под названием «универсальный индикатор», окраска которого меняется от ярко-красной в сильнокислой среде до жѐлтой в нейтральном растворе и сине-зелѐной в растворе щѐлочи. Измеряемые значения рН обычно лежат в диапазоне 0-14. Пропитанную универсальным индикатором фильтровальную бумажку смачивают в растворе, рН которого нужно определить, а потом сравнивают цвет бумажки с заранее приготовленной цветовой шкалой, где каждый оттенок цвета соответствует определѐнному значению рН. Более точные значения рН определяют электрометрическим методом, измеряя электропроводность раствора специальными приборами – рН – метрами или иономерами
(Рис. 14.1).
Рис. 14.1. Измерение рН электрометрическим методом
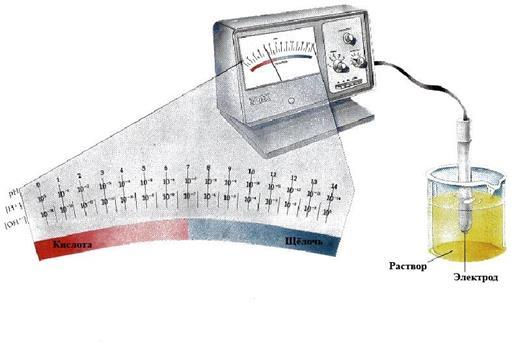
Принцип работы рН-метров основан на зависимости потенциала водородного электрода от концентрации водородных ионов в растворе
(см. Лекцию 4).
Однако кислую или щелочную реакцию могут иметь не только растворы кислот и щелочей, но и растворы солей вследствие гидролиза
.
14.3. Гидролиз
Гидролизом называется взаимодействие вещества с водой. Важнейшим случаем гидролиза является гидролиз солей. Можно сказать, что при гидролизе происходит разложение солей водой. Гидролизу подвергаются соли слабых кислот и оснований. Соли, образованные сильными кислотами и основаниями, гидролизу не подлежат. Реакции гидролиза следует писать в ионном виде, так как молекулярное уравнение не дает представления о происходящих при гидролизе соли процессах.
При гидролизе соли, образованной слабой кислотой и сильным основанием, реакция раствора будет щелочной. Гидролиз протекает по аниону
.
СН3
СООNa + H2
O ↔ CH3
COOH + NaOH;
СН3
СОО -
+ Na+
+ HOH ↔CH3
COOH + Na+
+ OH-
; или CH3
COO-
+ HOH ↔ CH3
COOH + OH-
;
При гидролизе соли, образованной сильной кислотой и слабым основанием, реакция раствора будет кислой. Гидролиз протекает по катиону
.
NH4
Cl + H2
O ↔ NH4
OH + HCl; NH4
+
+ Cl-
+ HOH ↔ NH4
OH + H+
+ Cl; или NH4
+
+ HOH ↔ NH4
OH + H+
;
Чем слабее кислота или основание, тем в большей степени подвергаются гидролизу (разлагаются водой) их соли.
Гидролиз солей, образованных слабой многоосновной кислотой или основанием многовалентного металла идет ступенчато: Na2
CO3
+ H2
O ↔ NaHCO3
+ NaOH;
1-я ступень: CO3
2-
+ HOH ↔ HCO3
-
+ OH-
; 2-я ступень: HCO3
- + HOH ↔ H2
CO3
+ OH-
;
Поскольку первая константа диссоциации всегда больше последующих, гидролиз по первой ступени всегда протекает в большей степени, чем гидролиз по второй и третьей ступени.
Аналогично происходит гидролиз соли, образованной основанием многовалентного металла:
СuCl2
+ H2
O ↔ СuOHCl + HCl; 1-я ступень: Сu2+
+ HOH ↔ CuOH+
+ H+
; 2-я ступень: CuOH+
+ HOH ↔ Cu(OH)2
+ H+
;
Гидролиз по второй ступени протекает в ничтожно малой степени.
Особенно глубоко протекает гидролиз солей, образованных слабым основанием и слабой кислотой. Гидролиз катиона и гидролиз аниона взаимно усиливают друг друга, смещая соответствующее равновесие вправо.
Аl(CH3
COO)3
+ H2
O → Al(OH)3
+ 3CH3
COOH; Al3+
+ HOH ↔ AlOH2+
+ H+
; AlOH2+
+ HOH ↔ Al(OH)2
+
+ H+
; Al(OH)2
+ + HOH ↔ Al(OH)3
+ H+
; 3СH3
COO -
+3HOH ↔ 3CH3
COOH + 3OH-
; 3H+
+ 3OH-
↔ 3H2
O
Ацетат алюминия гидролизуется необратимо и полностью, образуя малорастворимое соединение - гидроксид алюминия и малодиссоциированное соединение - уксусную кислоту. Ионы водорода и гидроксила образуют молекулы слабого электролита – воды, которая уходит из сферы реакции, тем самым, усиливая гидролиз катиона алюминия по второй и третьей ступени.
При смешении растворов солей, образованных слабой кислотой и сильным основанием и слабым основанием и сильной кислотой, гидролиз также взаимно усиливается. Так, при смешении растворов солей азотнокислого железа (III) и углекислого натрия выпадает осадок гидроксида железа (III) и выделяется углекислый газ вследствие разложения слабой угольной кислоты, образующейся вследствие гидролиза.
2Fe(NO3
)3
+ 3Na2
CO3
+ 3H2
O → 2Fe(OH)3
+ 3CO2
+ 6NaNO3
Fe3+
+ HOH ↔ Fe(OH)2+
+ H+
; Fe(OH)2+
+ HOH ↔ Fe(OH)2
+
+ H+
; Fe(OH)2
+
+ HOH ↔ Fe(OH)3
+ H+
; СO3
2-
+ HOH ↔ HCO3
-
+ OH-
; HCO3
-
+ HOH ↔ H2
CO3
+ OH-
;
Гидролиз отдельно взятых нитрата железа (III) и карбоната натрия ограничивается первой ступенью, однако при смешении растворов образующиеся ионы водорода и гидроксила связываются в молекулу слабого электролита – воды, который уходит из сферы реакции. При этом равновесие сильно смещается вправо, гидролиз каждой из солей усиливается и идет до полного разложения обеих солей.
С гидролизом солей приходится считаться и в лабораторной, и в производственной практике. Подавить нежелательный гидролиз можно, добавляя кислоту в растворы, имеющие, вследствие гидролиза, кислую реакцию, и щелочь в растворы со щелочной реакцией. Гидролиз является обратимым процессом, поэтому равновесие в растворах солей, подверженных гидролизу, можно сместить в ту или иную сторону. Константа гидролиза Кг
для солей слабой кислоты совпадает с константой диссоциации этой кислоты:
H
n
K
г
 (14.6) (14.6)
а константа гидролиза для солей слабого основания – с константой его диссоциации:
Me
n
(14.7)
Степень гидролиза связана с константой гидролиза соотношением:
(14.8)
где К – константа гидролиза, а С – концентрация раствора соли.
14.4. Буферные растворы
Часто в ходе какого-либо химического процесса необходимо поддерживать рН раствора на постоянном уровне. С этой целью в раствор буферные смеси
. Это водные растворы кислот и сопряжѐнных с ними оснований, например, уксусной кислоты и ацетата натрия или аммиака (гидроксида аммония) и хлорида аммония. Значение рН такой смеси остаѐтся постоянным
даже при сильном разбавлении или при добавлении в буферный раствор сильной кислоты или щѐлочи, то есть буферный раствор обладает определѐнной буферной ѐмкостью
. Буферной ѐмкостью называется количество моль-эквивалентов кислоты или щелочи, при добавлении которых к 1 л буферного раствора его рН изменится на 1 единицу. Рассмотрим механизм действия буферной смеси на примере ацетатного буферного раствора. Уксусная кислота в растворе диссоциирует по схеме:
CH3
COOH ↔ CH3
COO-
+ H+
(14.8)
Равновесие в данном процессе сильно сдвинуто вправо, так как уксусная кислота – слабый электролит; константа еѐ диссоциации
n
 определяется соотношением: K
д определяется соотношением: K
д
 1,754 10 5 1,754 10 5
Степень диссоциации уксусной кислоты понижается при добавлении в раствор ацетата натрия, который, будучи сильным электролитом, нацело диссоциирует по схеме:
CH3
COONa → CH3
COO-
+ Na+
Ацетат-анионы, попадая в раствор, сдвигают равновесие процесса (14.8) влево. Чем выше концентрация соли в растворе, тем меньше степень диссоциации кислоты и ниже концентрация водородных ионов. Меняя концентрации кислоты и еѐ соли в растворе, можно получать растворы с различным значением рН. Рассчитать рН буферного раствора можно по следующему соотношению:
 pH pK
д
lg pH pK
д
lg (14.9) (14.9)
где pK
д
- отрицательный логарифм константы диссоциации кислоты HA, а
[НА] и [А-
] – молярные концентрации кислоты и аниона кислоты в растворе.
В ацетатном буферном растворе рН обычно устанавливают равным 4,73. Если к такому раствору прибавить сильную кислоту, то поступающие в раствор вследствие полной диссоциации сильной кислоты катионы водорода полностью связываются присутствующими в растворе ацетат-анионами (равновесие 14.8 сдвигается влево) в молекулы слабой уксусной кислоты, поэтому концентрация водородных ионов в буферном растворе практически не изменяется. Так, если к 1л ацетатной буферной смеси прибавить 100 мл раствора НCl с концентрацией 0,1М, то рН буферного раствора изменится всего на 0,1 (с 4,73 до 4,64), тогда как при добавлении такого же количества кислоты к 1 л чистой воды приведѐт к изменению рН от 7 до 2.
Прибавленная в буферную смесь щѐлочь будет реагировать с молекулами уксусной кислоты по схеме:
CH3
COOH + NaOH→ CH3
COONa + H2
O
При этом равновесие диссоциации уксусной кислоты сместится вправо, однако повышение концентрации ацетат-аниона приведѐт к снижению степени диссоциации кислоты, поэтому рН буферного раствора и в этом случае изменится незначительно. При добавлении к 1л ацетатной буферной смеси 100 мл раствора NaOH с концентрацией 0,1М рН буферного раствора изменится меньше чем на 0,1 (с 4,73 до 4,82), тогда как при добавлении такого же количества кислоты к 1 л чистой воды рН возрастѐт с 7 до 12.
Способность буферных смесей поддерживать постоянство рН не безгранична. Если количество прибавленной кислоты или щѐлочи превысит буферную ѐмкость, то рН раствора резко изменится.
14.5. Окислительно-восстановительные процессы
В молекулах сложных веществ осуществляются, как правило, полярные ковалентные связи. Неравномерность распределения электронов в молекулах, состоящих из атомов с различной электроотрицательностью, называется окисленностью
. Менее электроотрицательный элемент, электроны которого смещаются к более электроотрицательному элементу, приобретает положительную
окисленность, а более электроотрицательный элемент - отрицательную
окисленность. В ионных соединениях валентные электроны полностью переходят от одного атома, который становится положительно заряженным катионом
, к другому, становящемуся отрицательно зараженным анионом
.
Число электронов, смещенных от
одного атома данного элемента (при положительной окисленности) или к
одному атому данного элемента (при отрицательной окисленности) называется степенью окисленности
элемента, или степенью окисления
, или окислительным числом
. Наиболее употребительным является термин «степень окисления». Для большинства химических соединений понятие степени окисления является условным, так как не отражает реальный заряд данного атома, однако это понятие широко используется в химии и помогает понять природу многих химических процессов.
В простых веществах
степень окисления всегда равна нулю.
Некоторым элементам свойственна одна степень окисления, другие проявляют различные степени окисления в различных соединениях. Постоянную степень окисления, (+1) имеют щелочные металлы первой группы и щелочноземельные металлы второй группы (+2). Фтор всегда имеет степень окисления (-1). Для водорода степень окисления в большинстве соединений равна (+1), а для кислорода, как правило (-2). Зная формулу химического соединения, можно определить степень окисления любого элемента, если в состав соединения входят вышеупомянутые элементы с постоянной степенью окисления, учитывая, что молекула в целом должна быть электронейтральной.
В качестве примера рассчитаем степень окисления азота в следующих соединениях:
NH3
: х + 3(+1) = 0; х = -3; NaNO2
: +1 + х + 2(-2) = 0; х =+3; KNO3
: +1 + х + 3(-2) = 0; х = +5
.
Иногда расчет оказывается затруднительным. Например, в соединениях PI3
, NI3
для определения степени окисления элементов следует использовать значения электроотрицательности элементов. Так, электроотрицательности фосфора, йода и азота равны, соответственно, 2,2; 2,6 и 3,07. Учитывая, что при образовании химической связи электроны смещаются в сторону элемента с большей электроотрицательностью, определяем степени окисления фосфора и йода в йодиде фосфора как (+3) и (-1) соответственно, а азота и йода в йодиде азота как (-3) и (+1) соответственно.
Необходимо также уметь рассчитывать заряд ионов, входящих в состав молекулы. Например, молекула серной кислоты H2
SO4
распадается в растворе на ионы Н+
и SO4
-2
; молекула хлорида натрия NaCl
- на ионы Na+
и Cl-
; молекула дихромата калия K2
Cr2
O7
- на ионы K+
и Сr2
O7
-
.
Нерастворимые соединения не образуют ионов и записываются в виде нейтральных молекул. В виде нейтральных молекул записываются и газы.
При окислительно-восстановительных реакциях электроны переходят от атомов одних элементов к атомам других элементов. Вещества, присоединяющие электроны, называются окислителями
, а процесс присоединения электронов называется восстановлением.
Следовательно, окислитель
в окислительно-восстановительной реакции восстанавливается
.
Вещества, теряющие электроны, называются восстановителями
, а процесс отдачи электронов называется окислением
. Следовательно, восстановитель
в процессе окислительно-восстановительной реакции окисляется
.
Металлы проявляют в своих соединениях только положительную окисленность, и низшая степень их окисления (в свободном состоянии) равна нулю. Следовательно, все свободные металлы являются восстановителями. Многие металлы имеют несколько степеней окисления. Соединения металлов в низшей степени окисления служат сильными восстановителями. Это, например, соли железа (II), олова (II), хрома (III), меди (I). Сильными окислителями являются соединения металлов в высшей степени окисления (равной номеру группы), например, соли железа (III), ртути (II), марганца (VII), хрома (VI).
Соединения неметаллов в положительных высших степенях окисления являются сильными окислителями, например, соединения азота (V), серы (VI), хлора (VII). Соединения, в которых неметалл проявляет низшую степень окисления, служат сильными
восстановителями, например, соединения азота в степени окисления (-3). Соединения элементов в промежуточных степенях окисления (например, азота(III)) могут служить как окислителями, так и восстановителями, в зависимости от того, реагируют ли они с более сильным окислителем или с более сильным восстановителем.
Из простых веществ в качестве окислителей чаще всего применяются галогены (окислительная способность галогенов с увеличением порядкового номера снижается) и кислород, а в качестве восстановителя углерод или его моноксид СО.
Сила кислородсодержащих кислот растѐт с возрастанием степени окисления центрального атома кислоты. Например, азотная кислота HNO3
(степень окисления азота равна +5) сильнее азотистой HNO2
(соответственно +3), а серная кислота H2
SO4
(степень окисления серы равна +6) сильнее сернистой H2
SO3
(соответственно +4).
Составление уравнений окислительно-восстановительных реакций и подбор коэффициентов в них производится двумя методами, основной которых служит принцип сохранения заряда: в окислительновосстановительных реакциях общее число электронов, отдаваемых восстановителем, равно общему числу электронов, присоединяемых окислителем.
В методе электронного баланса
подсчет числа присоединяемых и теряемых электронов производится на основании значений степеней окисления элементов до начала и после окончания реакции. Например, в реакции
KMnO4
+ FeSO4
+ H2
SO4
= MnSO4
+ Fe2
(SO4
)3
+ K2
SO4
+ H2
O
Степень окисления изменяют только марганец и железо. Записываем уравнение электронного баланса:
Mn+7
+ 5e = Mn+2
1 Fe+2
- 1e = Fe+3
5 _______________________ Mn+7
+ 5Fe+2
= Mn+2
+ 5Fe+3
Таким образом, коэффициенты в уравнении реакции при окислителе и восстановителе - это 1 и 5. Однако, следует учесть, что в результате реакции образуется Fe2
(SO4
)3
, содержащий 2 моля Fe (III),поэтому коэффициенты следует удвоить. В итоге получаем следующее уравнение:
2KMnO4
+ 10FeSO4
+ H2
SO4
= 2MnSO4
+ 5Fe2
(SO4
)3
+ K2
SO4
+ H2
O
Остальные коэффициенты находят по балансу других элементов (пока без кислорода и водорода), то есть в данном случае, калия и серы:
2KMnO4
+ 10FeSO4
+ 8H2
SO4
= 2MnSO4
+ 5Fe2
(SO4
)3
+ K2
SO4
+ H2
O
Далее по балансу атомов водорода определяют число молей воды
2KMnO4
+ 10FeSO4
+ 8H2
SO4
= 2MnSO4
+ 5Fe2
(SO4
)3
+ K2
SO4
+ 8H2
O
Для проверки правильности подобранных коэффициентов сводят баланс атомов кислорода: слева 8+40+32=80 атомов кислорода; справа также 8+60+4+8=80 атомов кислорода. Следовательно, коэффициенты определены правильно.
Ионно-электронный метод
основан на составлении частных уравнений восстановления ионов (молекул) окислителя и окисления ионов (молекул) восстановителя. Для этого необходимо составить ионную схему реакции (ионы, не содержащие атомов, изменяющих степень окисления, в схему не включаются).
Вернемся к ранее рассмотренному примеру:
KMnO4
+ FeSO4
+ H2
SO4
= MnSO4
+ Fe2
(SO4
)3
+ K2
SO4
+ H2
O
Как видим, ионы Fe+2
окисляются до Fe+3
, а ионы MnO4
-
превращаются в ионы Mn+2
. Частные уравнения окисления восстановителя и восстановления окислителя имеют вид:
Fe+2
- 1e = Fe+3
MnO4
-
+ 8H+
+ 5e = Mn+2
+ 4H2
O
Для того чтобы связать атомы кислорода в воду, необходимо добавить двукратное количество атомов водорода (так как реакция идет в кислой среде). Уравнивая количество отданных и полученных электронов, получаем ионное уравнение:
| 2Fe
+2 - 2e = 2Fe
+3
|
5
|
| MnO4
-
+ 8H+
+ 5e = Mn+2
+ 4H2
O
|
2
|
___________________________________________ 10Fe+2
+ 2MnO4
-
+16H+
= 10Fe+3
+ 2Mn+2
+ 8H2
O
Окончательное молекулярное уравнение имеет вид:
2KMnO4
+ 10FeSO4
+ 8H2
SO4
= 2MnSO4
+ 5Fe2
(SO4
)3
+ K2
SO4
+ 8H2
O
При использовании ионно-электронного метода все коэффициенты получаются сразу, поэтому при составлении уравнений реакций, идущих в кислой, щелочной и нейтральной среде этот метод предпочтительнее. В нейтральной среде для прибавления и отнятия кислорода используют молекулы воды, в щелочной среде - ионы гидроксила и воды.
Примеры окислительно-восстановительных реакций:
1) K2
Cr2
O7
+ H2
SO4
+ Na2
SO3
→ Cr2
(SO4
)3
+ Na2
SO4
+ K2
SO4
+ H2
O
Cr2
O7
-2
+ 14H+
+ 6e = 2Cr+3
+ 7H2
O 1
SO3
-2
+ H2
O -2e = SO4
-2
+ 2H+
3
Cr2
O7
-2
+ 3SO3
-2
+ 18H+
= 2Cr+3
+ 3SO4
-2
+ 4H2
O
K2
Cr2
O7
+ 4H2
SO4
+ 3Na2
SO3
= Cr2
(SO4
)3
+ 3Na2
SO4
+ K2
SO4
+ 4H2
O
2) K2
Cr2
O7
+ H2
SO4
+ NaNO2
→ Cr2
(SO4
)3
+NO2
+Na2
SO4
+K2
SO4
+ H2
O
Cr2
O7
-2
+ 14H+
+ 6e = 2Cr+3
+ 7H2
O 1
NO2
-
- 1e = NO2
0
6
Cr2
O7
-2
+ 14H+
+ 6NO2
-
= 2Cr+3
+ 6NO2
+ 7H2
O
K2
Cr2
O7
+ 7H2
SO4
+ 6NaNO2
= Cr2
(SO4
)3
+6NO2
+ 3Na2
SO4
+ K2
SO4
+ 7H2
O 3) K2
Cr2
O7
+ H2
SO4
+ KI → Cr2
(SO4
)3
+ I2
+ K2
SO4
+ H2
O
Cr2
O7
-2
+ 14H+
+ 6e = 2Cr+3
+ 7H2
O 1
2I-
- 2e = I2
0
3
Cr2
O7
-2
+ 14H+
+ 6I-
= 2Cr+3
+ 3I2
+ 7H2
O
K2
Cr2
O7
+ 7H2
SO4
+ 6KI = Cr2
(SO4
)3
+ 3I2
+ 4K2
SO4
+ 7H2
O
4) KMnO4
+ Na2
SO3
+ H2
SO4
→ MnSO4
+ Na2
SO4
+ K2
SO4
+ H2
O
MnO4
-2
+ 8H+
+ 5e = Mn+2
+ 4H2
O 2
SO3
-2
+ H2
O - 2e = SO4
-2
+ 2H+
5
2MnO4
-2
+ 6H+
+ 5SO3
-2
= 2Mn+2
+ 3H2
O + 5SO4
-2
2KMnO4
+ 5Na2
SO3
+ 3H2
SO4
= 2MnSO4
+ 5Na2
SO4
+ K2
SO4
+ 3H2
O
5) KMnO4
+ Na2
SO3
+ KOH → K2
MnO4
+ Na2
SO4
+ H2
O
MnO4
-
- 1e = MnO4
-2
2
SO3
-2
+2OH-
+2e = SO4
-2
+ H2
O 1
2MnO4
-
+ SO3
-2
+ 2OH-
= 2MnO2
-2
+ SO4
-2
+ H2
O
2KMnO4
+ Na2
SO3
+ 2KOH = 2K2
MnO4
+ Na2
SO4
+ H2
O
6) KMnO4
+ Na2
SO3
+ H2
O → MnO2
+ Na2
SO4
+ KOH
MnO2
-
+ 4H2
O + 3e + MnO2
+ 4OH-
+ 2H2
O 2
SO3
-2
+ 2OH-
- 2e = SO4
-2
+ H2
O 3
2MnO2
-
+ H2
O + 3SO3
-2
= 2MnO2
+ 2OH-
+ 3SO4
-2
2KMnO4
+ 3Na2
SO3
+ H2
O = 2MnO2
+ 3Na2
SO4
+ 2KOH
7) KI + KIO3
+ H2
SO4
→ I2
+ K2
SO4
+ H2
O
2I-
- 2e = I2
0
5
2IO3
-
+ 12H+
+ 10e = I2
+ 6H2
O 1
10I-
+ 2IO3
-
+ 12H+
= 5I2
+ I2
+ 6H2
O
10KI + 2KIO3
+ 6H2
SO4
= 6I2
+ 6K2
SO4
+ 6H2
O
5KI + KIO3
+ 3H2
SO4
= 3I2
+ 3K2
SO4
+ 3H2
O
8) KI + KMnO4
+ H2
SO4
→ I2
+ MnSO4
+ K2
SO4
+ H2
O
2I-
- 2e = I2
0
5
MnO4
-
+ 8H+
+ 5e = Mn2+
+ 4H2
O 2
10I-
+ 2MnO2
-
+ 16H+
= 5I2
+ 2Mn2+
+ 8H2
O
10KI + 2KMnO4
+ 8H2
SO4
= 5I2
+ 2MnSO4
+ 6K2
SO4
+ 8H2
O
9) KI + NaNO2
+ H2
SO4
= I2
+ NO + K2
SO4
+ Na2
SO4
+ H2
O
2I-
- 2e = I2
0
1
NO2
-
+ 2H+
+1e = NO + H2
O 2
2I- + 2NO2-
+ 4H+
= I2
+ NO + H2
O
2KI +2NaNO2
+ 2H2
SO4
= I2
+ 2NO + K2
SO4
+ Na2
SO4
+ 2H2
O
Реакции самоокисления-самовосстановления
, когда степень окисления одного и того же элемента и повышается, и понижается, называются реакциями диспропорционирования
. Примером может служить реакция хлора с гидроксидом калия:
2Cl2
+ 6KOH = 5KCl + KClO3
+ 3H2
O Cl0
- 5e = Cl+5
1 Cl0
+ 1e = Cl-
5 3Cl2
= Cl+5
+ 5Cl-
В этой реакции хлор выступает и как восстановитель, и как окислитель.
14.6. Вопросы и задания:
1. Что такое кислота и основание с точки зрения теории электролитической диссоциации и с точки зрения протонной теории?
2. Какой из растворов является раствором слабого электролита: уксусной кислоты, гидроксида натрия, гидроксида аммония, хлорида натрия, дихромата калия, соляной кислоты?
3. Сколько молей соляной кислоты потребуется для приготовления 2 л раствора с рН=1? Сколько молей гидроксида натрия NaOH содержится в 5 л раствора, рН которого равен 13?
4.   Какое соотношение верно для нейтральных растворов: pH
> pOH
; pH
< pOH
; pH pOH
7; pH pOH
14? Какое соотношение верно для нейтральных растворов: pH
> pOH
; pH
< pOH
; pH pOH
7; pH pOH
14?
5. Какие из солей: Al2
(SO4
)3
; K2
S, Pb(NO3
)2
; NaCl, NH4
Cl, Na2
CO3
, K2
SO4
– подвергаются гидролизу? Составьте ионные уравнения гидролиза соответствующих солей.
6. Какая из следующих солей: CuSO4
, Na2
SiO3
, KNO3
, NH4
Cl, CH3
COONa, SnCl2
, Na2CO3 подвергается гидролизу по катиону? По аниону?
7. Определите пропущенные соединения в реакции: Al2
S3
+ H2
O→ … + …
8. Что такое степень окисления, степень окисленности, окислительное число, окисленность?
9. Какие реакции называются окислительно-восстановительными?
10. Какие вещества могут быть только окислителями, какие вещества могут быть только восстановителями, а какие могут проявлять как свойства окислителя, так и свойство восстановителя?
11. Вычислите степень окисления азота в аммиаке NH3
, нитрите натрия NaNO2
, диоксиде азота NO2
, азотной кислоте HNO3
.
12. Вычислите степени окисления хлора во всех его кислородных кислотах HClO, HClO2
, HClO3
и HClO4
. Какая из этих кислот является самым сильным
окислителем? Какая кислота самая сильная?
13. Какие коэффициенты нужно поставить перед восстановителем и окислителем в реакциях: K2
Cr2
O7
+ H2
SO4
+ NaNO2
→ Cr2
(SO4
)3
+ NO2
+ Na2
SO4
+ K2
SO4
+ H2
O; KMnO4
+ Na2
SO3
+ H2
SO4
→ MnSO4
+ Na2
SO4
+ K2
SO4
+ H2
O; C + H2
SO4
(к) → CO2
+ SO2
+ H2
O?
14. Составьте электронно-ионные схемы и на их основе расставьте коэффициенты в следующих окислительно-восстановительных реакциях: а) P + HNO3
+ H2
O → H3
PO4
+ NO; б) H2
S + HNO3
→ S + NO2
+ H2
O;
в) Hg + H2
SO4
→ HgSO4
+ SO2
+ H2
O; г) FeCl3
+ HI → FeCl2
+ HCl + I2
;
д) H2
S + SO2
→ S + H2
O
15. Какие вещества восстанавливаются и окисляются в следующих реакциях? а) 2KI + 2FeCl3
→
2FeCl2
+ I2
+ 2KCl; б) 3Cl2
+ 6KOH →
KClO3
+ 5KCl + 3H2
O; в) Ag + 2HNO3
→
AgNO3
+ NO2
+ H2
O; г) Pb(NO3
)2
→
PbO +
NO2
+ O2
; д) H2
S + SO2
→ S + H2
O
Лекция 15. Химическая связь; комплиментарность
Химические свойства элемента обусловлены способностью его атома терять и приобретать электроны. Эта способность количественно оценивается энергией ионизации атома
и его сродством к электрону
.
15.1. Энергия ионизации и сродство к электрону
Энергией ионизации называется количество энергии, необходимое для отрыва электрона от невозбужденного атома и выражается в килоджоулях (Кдж)/моль или электрон-вольтах (эВ)/атом. Для многоэлектронных атомов энергия, необходимая для отрыва каждого последующего электрона всегда больше, чем энергия для отрыва предыдущего электрона, так как отрывать электрон приходится не от нейтрального атома, а от положительно заряженного иона (ионом называется атом, потерявший или приобретший один или несколько электронов)
. Энергия ионизации атома зависит от его электронной конфигурации. Наименьшими значениями энергии ионизации обладают s - элементы первой группы, наибольшими - элементы восьмой группы.
Сродством к электрону
называют энергетический эффект присоединения электрона к нейтральному атому, который превращается при этом в отрицательно заряженный ион (его выражают через энергию ионизации отрицательных ионов). Наибольшим сродством к электрону обладают р - элементы седьмой группы. Наименьшее и даже отрицательное сродство к электрону имеют инертные газы и элементы второй группы (конфигурации s2
и s2
p2
). Высоким сродством к электрону обладают кислород, сера, углерод и некоторые другие элементы. Электроотрицательностью
называют способность атома данного элемента к оттягиванию на себя электронной плотности по сравнению с другими элементами, входящими в соединение. Эта способность зависит от энергии ионизации атома и его сродства к электрону (иногда ее считают равной полусумме того и другого
). Большое значение имеет атомный радиус
(атомы и ионы не имеют строго определенных границ вследствие волновой природы электрона, поэтому определяют условные радиусы атомов и ионов, связанных химической связью в кристаллах). Радиусы атомов в периодах с ростом порядкового номера уменьшаются, так как возрастает заряд ядра, а, следовательно, и притяжение к нему электронов. В пределах одной группы атомные радиусы возрастают, так как возрастает число энергетических уровней (электронных слоев).
Таблица 15.1. Относительные электроотрицательности некоторых атомов в виде полусуммы сродства к электрону и потенциала ионизации, эВ
| Период
|
Группы элементов
|
| I
|
II
|
III
|
IV
|
V
|
VI
|
VII
|
VIII
|
| 1
|
Н
2,1
|
He
|
| 2
|
Li
0,97
|
Be 1,47
|
B 2,01
|
C 2,50
|
N 3,07
|
O
3,50
|
F 4,10
|
Ne
|
| 3
|
Na 1,01
|
Mg
1,23
|
Al 1,47
|
Si 1,47
|
P 2,1
|
S 2,6
|
Cl 2,83
|
Ar
|
| 4
|
K 0,91
|
Ca 1,04
|
Sc 1,20
|
Ti
1,32
|
V 1,45
|
Cr 1,56
|
Mn
1,60
|
Fe
1,64
Co
1,70
Ni
1,75
|
| 5
|
Cu
1,75
Rb
0,89
|
Zn
1,66
Sr
0,99
|
Ga
1,82
|
Ge
2,02
|
As 2,20
|
Se 2,48
|
Br 2,74
|
Kr
|
Электроотрицательность элементов периодической системы, как правило, последовательно возрастает слева направо в каждом периоде. В пределах каждой группы, за несколькими исключениями, электроотрицательность последовательно убывает сверху вниз. Электроотрицательность атома зависит и от степени его окисления. Так, для трех оксидов хрома наблюдается изменение свойств от основного характера (CrO) через амфотерный (Сr2
O3
) до кислотного (СrO3
). Один и тот же элемент хром в соединении СrO ведет себя как типичный металл, в Сr2
O3
– как амфотерный металл, а в СrO3
как типичный неметалл.
При составлении химических формул соединений более электроотрицательные элементы помещаются правее, чем менее электроотрицательные, например, H2
S, OF2
, SCl2
O, Br3
N, SiBr2
F2
Химическая связь возникает при электромагнитном взаимодействии атомов с образованием химически устойчивой двух- или многоатомной системы (молекулы
). Химическая связь образуется только в том случае, если при сближении двух или более атомов полная энергия системы (сумма кинетической и потенциальной энергии) понижается. При сближении двух атомов между ними возникают электростатические силы двух типов - сила притяжения электрона к обоим ядрам и сила отталкивания между ядрами (и электронами). Молекула образуется в том случае, если равнодействующая сил притяжения и отталкивания равна нулю, то есть взаимное отталкивание ядер должно быть скомпенсировано притяжением электронов к ядрам. Химическая связь образуется в том случае, если электрон оказывается между ядрами.
15.2. Валентность
Валентностью элемента называют способность его атома к образованию химической связи.
Это свойство атомов одного элемента соединяться с определенным количеством атомов другого элемента или замещать их. Количественной мерой валентности принято считать число различных атомов в молекуле, с которыми данный атом образует связи. Теория валентных связей
локализованных электронных пар (ВС) исходит из положения, что одна химическая связь образуется двумя неспаренными электронами с антипараллельными спинами.
При этом происходит обобществление электронов и образуется электронная пара, принадлежащая одновременно двум атомам.
Спаренные электроны на атомных орбиталях могут при возбуждении их разъединяться (распариваться
) при наличии свободных ячеек того же уровня. Распаривание электронов на другой уровень невозможно. Например, валентность элементов первой группы равна 1, так как на внешнем уровне эти атомы имеют один электрон. Валентность элементов второй группы в невозбужденном состоянии равна 0, так как на внешнем уровне у них нет неспаренных электронов. При возбуждении этих атомов спаренные электроны распариваются в свободные ячейки р - подуровня того же уровня, и их валентность становится равной 2. Кислород и фтор во всех соединениях проявляют постоянную валентность
, равную 2 для кислорода и 1 для фтора, так как валентные электроны этих элементов находятся на втором энергетическом уровне, где нет свободных квантовых ячеек. В то же время сера - аналог кислорода - проявляет переменную валентность
, равную 2, 4 и 6; хлор - аналог фтора - проявляет переменную валентность, равную 1, 3, 5 и 7. Это объясняется наличием на третьем энергетическом уровне свободных d - ячеек.
Валентная связь (пара электронов) схематически обозначается черточкой между двумя атомами, вступающими в связь. Например, двухатомная молекула водорода обозначается так: Н - Н;
молекула воды: Н - О - Н;
молекула кислорода: О = О
15.3. Виды химической связи
Химическая связь характеризуется следующими характеристиками:
Длина связи
равна расстоянию между ядрами атомов в молекуле.
Энергия связи
равна количеству энергии, выделяющемуся при образовании связи (так как полная энергии молекулы меньше суммы энергий составляющих ее атомов). Эта величина является важнейшей характеристикой прочности связи (и измеряется в кДж/моль).
Наиболее прочные химические связи возникают в направлении максимального перекрывания электронных облаков. Поскольку атомные орбитали имеют определенную форму, их максимальное перекрывание возможно при определенной пространственной ориентации области перекрывания электронных облаков. Поэтому химическая связь характеризуется направленностью
. Различают  (сигма), (сигма),
 (пи)
и (пи)
и  (дельта)
-связи. Сигма - связь
возникает при перекрывании орбиталей, направленных вдоль оси, соединяющей ядра взаимодействующих атомов (в простейшем случае двух s - орбиталей в молекуле водорода Н2
). Пи - связь
образуется при перекрывании атомных орбиталей, расположенных по обе стороны оси, соединяющей ядра атомов (в основном, при перекрывании облаков р-электронов). Дельта - связь
возникает при перекрывании всех четырех лопастей двух d - орбиталей, расположенных в параллельных плоскостях. При наложении пи - и дельта - связей на сигма -связи образуются двойные и тройные связи ( например, молекула этилена СН2
= СН2
; молекула О=С=О; молекула ацетилена СН (дельта)
-связи. Сигма - связь
возникает при перекрывании орбиталей, направленных вдоль оси, соединяющей ядра взаимодействующих атомов (в простейшем случае двух s - орбиталей в молекуле водорода Н2
). Пи - связь
образуется при перекрывании атомных орбиталей, расположенных по обе стороны оси, соединяющей ядра атомов (в основном, при перекрывании облаков р-электронов). Дельта - связь
возникает при перекрывании всех четырех лопастей двух d - орбиталей, расположенных в параллельных плоскостях. При наложении пи - и дельта - связей на сигма -связи образуются двойные и тройные связи ( например, молекула этилена СН2
= СН2
; молекула О=С=О; молекула ацетилена СН СН и т.д.). Число связей между атомами называется кратностью, или порядком
связи. СН и т.д.). Число связей между атомами называется кратностью, или порядком
связи.
Если общая электронная пара не смещается ни к одному из атомов, она называется ковалентной неполярной
. Такие связи образуют атомы, которые почти не отличаются по своей электроотрицательности. Это, например, молекулы водорода, кислорода, азота (состоящие из одинаковых атомов) или молекула кристаллического карбида кремния SiC, а также связи С-Н в органических молекулах.
Если взаимодействующие атомы имеют различную электроотрицательность, то общая пара электронов смещается к ядру более электроотрицательного атома. Такая связь называется ковалентной полярной
. Например, электроотрицательность фтора (4,0эВ) больше электроотрицательности водорода (2,1 эВ), поэтому в молекуле фтористого водорода общая электронная пара смещена в сторону фтора. Вследствие смещения электронной пары средняя электронная плотность у одного атома будет выше, чем у другого, и молекула будет полярной.
При больших различиях в электроотрицательности элементов возникает ионная связь
. Более электроотрицательный атом «отрывает» электронную пару и становится отрицательно заряженным ионом, тогда как менее электроотрицательный атом превращается в положительно заряженный ион. К типичным соединениям с ионной связью относятся соединения активных щелочных металлов с галогенами (например, CsF, CsCl, NaCl).
Химическую связь можно охарактеризовать при помощи понятия «электроотрицательность». Чем больше отличаются по электроотрицательности атомы, образующих химическую связь, тем
ближе эта связь к ионному типу. Если разность
электроотрицательностей атомов больше 2,1, то такие связи могут считаться чисто ионными (по другим данным, при разности электроотрицательностей 1,7 связь является ионной на 50%). Если разность электроотрицательностей атомов меньше 1,7, то связь считается ковалентной. Если разность электроотрицательностей атомов равна нулю, то химическая связь лишена ионного характера и является ковалентной.
При составлении химических формул более электроотрицательные элементы помещаются правее, например, H2
S, OF2
, SCl2
O, Br3
N, SiBr2
F2
Донорно-акцепторной
связью называется такая связь, когда один из входящих в молекулу атомов имеет неподеленную электронную пару, а другой - свободную квантовую ячейку. Примером такой связи может служить образование положительно заряженного иона аммония NH4
+
или комплексного (сложного) иона ВН4
-
, где донором
является азот (в молекуле аммиака), а акцептором
- катион водорода; или донором является бор в молекуле гидрида бора, а донором - гидрид-ион Н-
. Донорно-акцепторная связь образуется в комплексных, или координационных,
соединениях.
Если к раствору сульфата меди приливать раствор аммиака, то образующийся голубой осадок основной соли меди легко растворяется в избытке аммиака, окрашивая жидкость в интенсивный синий цвет. Прибавление к полученному раствору щелочи не вызывает образования осадка гидроксида меди Cu(OH)2
, следовательно, в растворе присутствует так мало ионов Cu2+
.
Что даже при большом содержании ионов гидроксила не достигается произведение растворимости, достаточное для осаждения нерастворимого гидроксида меди. Отсюда можно сделать заключение, что при реакции сульфата меди с раствором аммиака ионы меди вступают в реакцию с прибавленным аммиаком и образуют какие-то новые ионы, которые не дают нерастворимого гидроксида меди при добавлении щелочи. В то же время, сульфат-ионы не претерпевают изменения, так как при прибавлении к раствору хлорида бария тотчас же выпадает в осадок нерастворимый сульфат бария. Темно-синяя окраска раствора при добавлении избытка аммиака к раствору сернокислой меди приводит, как было установлено, к образованию ионов сложного состава (комплексных ионов) [Cu(NH3
)4
]2+
.
При испарении воды эти комплексные ионы связываются с ионами SO4
2
-
и из раствора выделяются синие кристаллы соединения, состав которого выражается формулой [Cu(NH3
)4
]SO4
. Таким образом, при взаимодействии сульфата меди с избытком аммиака происходит следующая реакция: СuSO4
+ 4NH3
= [Cu(NH3
)4
]SO4
Ионы, которые образуются путем присоединения к данному иону нейтральных молекул или ионов противоположного знака, называются комплексными ионами.
Соли, в состав которых входят такие ионы, называются комплексными солями.
Комплексные соединения составляют наиболее обширный и разнообразный класс неорганических веществ. Известны не только комплексные соли, но и комплексные кислоты, комплексные основания и комплексные неэлектролиты. Многие природные комплексные соединения – витамин В12
, хлорофилл и другие – играют важную роль в биологических и физиологических процессах.
Строение и свойства комплексных соединений объясняет координационная теория
, созданная в 1893 году швейцарским химиком, лауреатом Нобелевской премии Альфредом Вернером. В соответствии с координационной теорией, в молекуле любого комплексного соединения один из ионов, обычно заряженный положительно, занимает центральное место и называется комплексообразователем
. Вокруг него в непосредственной близости координировано некоторое число отрицательно заряженных ионов или нейтральных молекул. Их называют лигандами
, или аддендами
. Комплексообразователь и лиганды составляют комплексный ион и образуют внутреннюю координационную сферу
комплексного соединения, которую при написании формулы комплексных соединений заключают в квадратные скобки. Остальные ионы находятся на более далеком расстоянии от центрального иона, составляя внешнюю координационную сферу
.
Например, в комплексном соединении К2
[Cd(CN)4
]
комплексообразователем является ион кадмия Cd2+
,
а лигандами – ионы CN-
.
Ион кадмия и цианид-ионы образуют внутреннюю сферу комплексного соединения. Внешняя сфера состоит из ионов калия, заряженных положительно. Заряд комплексного иона равен суммарному заряду внутренней сферы и противоположен ему по знаку. Например, в комплексном соединении состава K3
[Fe(CN)6
]
заряд комплексного иона равен –3, так как суммарный заряд комплексного соединения всегда равен нулю. Заряд комплексообразователя равен по величине и противоположен по знаку алгебраической сумме зарядов всех остальных ионов. Заряд комплексообразователя – иона железа – в вышеназванном соединении равен +3. Известны и такие комплексные соединения, где заряд комплексообразователя равен 0, например, в комплексах [Ca(NH3
)6
]
или [Fe(CO)5
]
, где комплексообразователями служат нейтральные атомы кальция и железа, а лигандами молекулы аммиака и оксида углерода. Количество лигандов в комплексе называется координационным числом комплексообразователя
.
Способность элемента-комплексообразователя к образованию комплексных соединений зависит от строения внешнего электронного уровня атома элемента и определяется его положением в системе Менделеева. Как, правило, комплексообразователями являются атомы или, чаще, ионы металлов, имеющих достаточное количество свободных орбиталей.
Природу химических связей в комплексных соединениях объясняют, используя различные модели координационной теории – метод валентных связей, теорию кристаллического поля и метод молекулярных орбиталей.
В соответствии с теорией валентных связей, при образовании комплексов возникает ковалентная связь по донорно-акцепторному механизму. Комплексообразователи имеют вакантные орбитали, то есть играют роль акцепторов. Лиганды имеют неподеленные пары электронов и служат донорами в донорно-акцепторном механизме образования связей. Например, ион цинка Zn2+
(элемент № 30) имеет следующую конфигурацию внешнего электронного уровня: 3d10
4s0
4p0
. На внешнем уровне у цинка имеются четыре вакантных атомных орбитали – одна 4s и три 4р. При взаимодействии иона цинка с молекулами аммиака, три атома азота которых имеют неподеленные пары электронов, возникают ковалентные связи по донорноакцепторному механизму, в результате чего образуется комплексный ион:
[Zn2+
] + 4 NH3
= [Zn(NH3
)4
]2+
Координационное число комплексообразователя зависит от числа вакантных орбиталей комплексообразователя. В соответствии с теорией Вернера, заряд комплексообразователя (или его степень окисления) является основным фактором, влияющим на координационное число
(КЧ) комплексообразователя. Имеет место следующая зависимость:
Z (Заряд) +1 +2 +3 +4 КЧ 2 4 6 8
Метод валентных связей
позволяет предсказать состав и структуру комплекса. Однако этот метод не может объяснить такие свойства комплексных соединений, как прочность, цвет и магнитные свойства.
Теория кристаллического поля
рассматривает электростатическое взаимодействие между комплексообразователями и лигандами. Имеет место как электростатическое притяжение положительно заряженного комплексообразователя с отрицательно заряженными лигандами, так и отталкивание лигандов друг от друга. Наиболее устойчив комплекс, в котором силы притяжения максимальны, а силы отталкивания минимальны. Устойчивость комплекса повышается, если происходит расщепление внешних d-орбиталей комплекса на подуровни. Электроны dγ
(dz2
, dx2-y2
) и dε
(dxy
, dxz
и d-yz
)
могут иметь различную энергию. Это приводит к появлению цвета у комплексного иона и проявлению им магнитных свойств вследствие изменения числа неспаренных электронов. Например, оранжевокрасная окраска комплексной соли K3
[Fe(CN)6
]
обусловлена тем, что при поглощении соответствующего кванта света возможен переход электрона с орбитали dε
на орбиталь dγ.
Комплексная соль K2
[Zn(CN)4
]
бесцветна, так как в данном случае переход электрона с орбитали dγ
на dε
- орбиталь не может быть осуществлен. Теория кристаллического поля объясняет наличие или отсутствие цвета у комплексных ионов
Метод молекулярных орбиталей
объясняет наличие в комплексных ионах неспаренных электронов (комплексы, имеющие несколько неспаренных электронов, называются высокоспиновыми
, не имеющие неспаренных электронов- низкоспиновыми
) и, следовательно, магнитные свойства комплексных соединений. Молекулярные орбитали образуются, когда атомные орбитали комплексообразователя и лигандов близки по энергии и соответствующим образом ориентированы в пространстве. Это приводит к образованию связывающих, несвязывающих и разрыхляющих молекулярных орбиталей. Так, например, в комплексном ионе [Ni(NH3
)6
]2+
15 молекулярных орбиталей – 6 связывающих, 6 разрыхляющих и 3 несвязывающих, на которых располагаются 20 валентных электронов, два из которых не спарены.
В настоящее время принята рациональная номенклатура
, основанная на рекомендациях ИЮПАК (Международного союза по чистой и прикладной химии). При составлении названия комплексного соединения надо пользоваться следующими правилами:
1) Первым в именительном падеже называют анион, а потом в родительном падеже – катион, независимо от того, который из них является комплексным.
2) Перечисляют в порядке увеличения их сложности (или в алфавитном порядке): лиганды-анионы, лиганды-молекулы, лиганды-катионы, а затем указывают центральный атом комплексообразователя. Если центральный атом входит в состав комплексного катиона, то используют русское название элемента, а в скобках римскими цифрами указывают степень его окисления. Если же центральный атом входит в состав комплексного аниона, то употребляют латинское название этого элемента, после него римской цифрой обозначают степень его окисления, а в конце прибавляют суффикс –ат.
3) К названиям лигандов-анионов прибавляют окончание –о (Cl-
- хлоро-, CN-
- циано, СО3
2-
- карбонато- и т.д.). Названия нейтральных лигандов совпадают с названиями молекул, за исключением воды (акво), аммиака (аммин), оксида углерода (карбонил) и др.
4) Число лигандов, присоединенных к комплексообразователю, указывают приставками моно- (эта приставка обычно опускается), ди-, три-, тетра, пента-, гекса и т.д., образованными от соответствующих греческих числительных.
Примеры: [Zn(NH3
)4
]Cl2
– дихлорид тетраммин цинка; K3
[Fe(CN)6
] – гексацианоферрат (III) калия; K4
[Fe(CN)6
] – гексацианоферрат (II) калия; [Ni(CO)4
] – тетракарбонил никеля; Na[Al(OH)4
] – тетрагидроксоалюминат натрия; K2
[PtCl6
] – гексахлорплатинат (IV) калия; [Co(NH3
)5
Cl]Cl2
– дихлорид пентаамминохлорокобальта (III), [Cr(H2
O)5
Cl]Cl2
– хлорид пентааквохрома
(III).
По знаку электрического заряда комплексные ионы делят на катионные и анионные. Катионные комплексные ионы
образуются в результате координации полярных молекул (NH3
, H2
O
) вокруг положительного иона-комплексообразователя. Например,
[Ni(H2
O)6
](NO3
)2
– нитрат гексааквоникеля (II); [Ag(NH3
)2
]Cl
– хлорид диамминсеребра (I). Анионные комплексные ионы
образуются в результате координации вокруг положительного иона-
комплексообразователя отрицательных ионов-лигандов. Например, Na2
[Pt(CN)4
Cl2
]
– дихлоротетрацианоплатинат (IV) натрия; K2
[Zn(OH)4
]
– тетрагидроксоцинкат (II) калия.
Нейтральные комплексные ионы
образуются в результате координации вокруг нейтрального атома молекул. Например, [Ni(CO)4
]
– тетракарбонил никеля, [Fe(CO)5
]
– пентакарбонил железа.
Комплексные соединения ведут себя в растворе как сильные электролиты, то есть полностью диссоциируют на комплексные ионы (внутреннюю сферу) и противоионы (внешнюю сферу). Процесс диссоциации при растворении гексацианоферрата (III) калия идет по схеме:
K3
[Fe(CN)6
] = 3K+
+ [Fe(CN)6
]3-
Комплексные ионы в растворе являются устойчивыми и диссоциируют незначительно. Так, гексацианоферрат (III) диссоциирует по схеме: [Fe(CN)6
]3-
= Fe3+
+ 6CN-
;
Диссоциация комплексных ионов подчиняется закону действия масс и может быть охарактеризована соответствующей константой равновесия, которая называется константой нестойкости соответствующего комплексного иона:
 K
н
[Fe
3 ] [CN
3 ]6 (17.1) K
н
[Fe
3 ] [CN
3 ]6 (17.1)
[Fe
(CN
)6
]
Константа нестойкости случит мерой устойчивости комплексного иона в растворе. Например, Кн
для комплексных ионов [Ag(CN)2
]-
; [Cu(CN)4
]3-
; [Zn(CN)4
]2-
равны соответственно 1,0∙10-21
; 5∙10-31
и 1∙10-15.
Следовательно, самым устойчивым в водном растворе будет ион с наименьшей константой нестойкости, то есть тетрацианокупрат, а самым нестойким – тетрацианоцинкат с наибольшим значением константы нестойкости.
По степени влияния лигандов на электронную структуру центрального иона-комплексообразователя они располагаются в следующий ряд:
J-
≤ Br-
≤ Cl-
≤ OH-
≤ F-
≤ H2
O ≤ SCN-
≤ NH3
≤ NO2
-
≤ CN-
Лиганды, находящиеся в начале ряда, называются слабыми, а стоящие в конце ряда, сильными. Прочность комплекса возрастает с увеличением силы лиганда.
Процессы комплексобразования широко используются в промышленности. Способность веществ образовывать комплексные соединения используется для разработки эффективных методов получения чистых металлов из руд, для получения редких металлов, сверхчистых полупроводниковых материалов, лекарственных препаратов и красителей, очистки сточных вод, нанесения покрытий. Комплексные соединения различного состава широко применяются в лабораторной практике. Например, содержащий органические лиганды диметилглиоксим, или реактив Чугаева, служит для определения ионов никеля и палладия; динатриевая соль этилендиаминтетрауксусной кислоты (комплексон III, или трилон Б) образует комплексные соединения с двухвалентными металлами, что позволяет применять этот реагент для титриметрического (объемного) анализа.
Металлическая
связь возникает в кристаллах металлов, при этом электроны обобществляются, а в узлах кристаллической решѐтки находятся катионы металла. Химическая связь в твердых телах имеет свои особенности и описывается с позиций зонной теории
. Эта теория основывается на модели свободных электронов
. Валентные электроны в металлических кристаллах обобществляются (делокализируются), при этом образуется катионная «решетка», помещенная в так называемую электронную жидкость
. Энергия сцепления частиц определяется преобладанием кулоновского притяжения между электронами и катионами над энергией отталкивания электронов за счет их кинетической энергии и катионов за счет ионного взаимодействия, причем вклад последнего невелик. Зонная теория учитывает влияние поля решетки на поведение электронов. Вместо отдельных энергетических подуровней N несвязанных атомов в твердом теле имеются зоны энергетических подуровней, образующихся в результате расщепления при образовании N МО и N АО в сильном электрическом поле. Эти зоны обозначаются так же, как и подуровни отдельных атомов, то есть 1s, 2s, 3p и т.д. Максимальное число электронов, которое может разместиться в энергетических зонах, определяется принципом Паули и равно 2(2l+1)N. Между энергетическими зонами размещаются запрещенные зоны, где электрон находиться не может. Свойства твердых тел определяются положением разрешенных и запрещенных зон и порядком расположения в них электронов. Под валентной зоной
понимается разрешенная энергетическая зона, в которой размещаются валентные электроны. Следующая за ней разрешенная зона называется зоной проводимости
. Особенностью проводников
является перекрытие валентной зоны и зоны проводимости. Валентная зона частично заполнена электронами. При наложении электрического поля электроны приобретают дополнительную энергию и могут перемещаться в зону проводимости. У полупроводников
заполненная электронами валентная зона и зона проводимости не перекрываются, но близки по энергии (например, ширина запрещенной зоны для кремния или германия составляет 1 эВ). При наложении электрического поля, повышении температуры или под действием других факторов электроны в валентной зоне возбуждаются и переходят через запрещенную зону в зону проводимости. Для изоляторов-диэлектриков заполненная электронами валентная зона и зона проводимости не перекрываются. Ширина запрещенной зоны между ними достаточно велика и составляет величину порядка 4-10 эВ. Поэтому электроны из валентной зоны не могут перейти в зону проводимости. Зонная теория объясняет многие свойства кристаллов различных элементов.
Кристаллы – это твѐрдые вещества, в которых мельчайшие частицы (атомы, ионы или молекулы) «упакованы» в определенном порядке. В результате при росте кристаллов на их поверхности самопроизвольно возникают плоские грани, а сами кристаллы принимают разнообразную геометрическую форму. В зависимости от строения, кристаллы делят на ионные, ковалентные, молекулярные и металлические
.
Ионные кристаллы
построены из чередующихся катионов и анионов, которые удерживаются в определенном порядке силами электростатического притяжения и отталкивания. Каждый ион может удержать вокруг себя столько ионов противоположного знака, сколько поместится в окружающем его объѐме пространства с учетом размеров ионов. При этом силы притяжения и отталкивания должны быть уравновешены, и кристалл в целом должен оставаться электронейтральным. В результате образуются различные кристаллические структуры. Так, при взаимодействии катионов натрия Na+
(с радиусом 0,1 нм) и анионов хлора Cl–
(радиус 0,18 нм) образуется простейшая кубическая кристаллическая решетка, в которой вершины куба попеременно заняты ионами Na+
и Cl–
. (См. Рис. 15.1). Аналогично устроены кристаллы хлорида калия KCl, оксидов бария BaO и кальция
CaO, а также многих других веществ.
Рис. 15.1 Кубические кристаллы (a) галита (NaCl), (б) алмаза (С) и (в) флюорита (CaF2
)

Более сложно устроены кристаллические решетки CaF2
(См. Рис. 15.1) и многих других ионных соединений. В некоторых ионных кристаллах сложные многоатомные анионы могут соединяться в цепи, слои или образовывать трехмерный каркас, в полостях которого располагаются катионы. Так, например, устроены силикаты. Ионные кристаллы образует большинство солей неорганических и органических кислот, оксиды, гидроксиды, соли. В ионных кристаллах связи между ионами прочные, поэтому такие кристаллы имеют высокие температуры плавления (801°С для NaCl, 2627°С для СаО).
В ковалентных кристаллах в узлах кристаллической решетки находятся одинаковые или разные атомы, связанные ковалентными связями. Эти связи прочные и направлены под определенными углами. Такие решетки образуют атомы многих элементов, поэтому их часто называют атомными кристаллами. Типичным примером является алмаз; в его кристалле каждый атом углерода связан с четырьмя другими атомами, находящимися в вершинах тетраэдра (см. Рис. 15.1). Ковалентные кристаллы образуют бор, кремний, германий, мышьяк, сульфид цинка ZnS, оксиды кремния SiO2
, рения ReO3
и титана TiO2
, цианид меди (I) CuSCN и многие другие соединения. Поскольку между полярной ковалентной и ионной связью нет резкой границы, то не существует такой границы и между ионными и ковалентными кристаллами. Ковалентные кристаллы, как правило, твердые и тугоплавкие.
Молекулярные кристаллы
построены из изолированных молекул, между которыми действуют сравнительно слабые силы притяжения. В результате такие кристаллы имеют намного меньшие температуры плавления и кипения, твердость их низка. Большинство молекулярных кристаллов — это кристаллы органических соединений, типичным представителем таких кристаллов является нафталин. Молекулярные кристаллы образуют также некоторые простые вещества, например, галогены, азот N2
, кислород O2
, сера S8
, бинарные соединения типа воды H2
O, диоксида углерода CO2
и азота N2
O4
, металлоорганические соединения и некоторые комплексные соединения. К молекулярным кристаллам относятся кристаллы полимеров, а также кристаллы белков и нуклеиновых кислот. Особым случаем молекулярных кристаллов являются кристаллы отвердевших инертных газов, атомы которых можно назвать «одноатомными молекулами». У типичных молекулярных кристаллов низкие температуры плавления, большие коэффициенты теплового расширения, высокая сжимаемость, малая прочность. В обычных условиях большинство молекулярных кристаллов не проводит тока, это диэлектрики. Однако некоторые молекулярные кристаллы, например органические красители, обладают свойствами полупроводников. Прочность молекулярных кристаллов зависит от размеров и сложности молекул. Так, кристаллы гелия (радиус атома 0,12 нм) плавятся при –271,4°С (под давлением 30 атмосфер), а ксенона (радиус 0,22 нм) – при –111,8° С; кристаллы фтора плавятся при –219,6° С, а йода – при +113,6° С; метана СН4
– при –182,5° С, а триаконтана
С30
Н62
– при +65,8° С.
Рис 15.2. Пространственные решѐтки кристаллов

Металлические кристаллы
образуют чистые металлы и их сплавы. Такие кристаллы можно увидеть на изломе металлических деталей. Кристаллическая решетка металлов образована катионами, которые связаны подвижными электронами («электронным газом»). Такое строение обусловливает электропроводность, ковкость, высокую отражательную способность (блеск) кристаллов. Металлические кристаллы образуют разные структуры за счѐт различной упаковки атомов-шаров в пространстве. Щелочные металлы, хром, молибден, вольфрам и др. образуют объемно-центрированную кубическую решетку; медь, серебро, золото, алюминий, никель и др. – гранецентрированную кубическую решетку (в ней помимо 8 атомов в вершинах куба имеются еще 6, расположенные в центре граней); бериллий, магний, кальций, цинк и др. – так называемую гексагональную плотную решетку (в ней 12 атомов расположены в вершинах прямоугольной шестигранной призмы, 2 атома – в центре двух оснований призмы и еще 3 атома – в вершинах треугольника в центре призмы). На Рис. 15.2 показаны различные пространственные кристаллические решѐтки.
Водородная связь
образуется вследствие того, что поляризованный атом водорода (в одной молекуле) способен глубоко внедряться в электронную оболочку соседнего отрицательно поляризованного атома (другой молекулы). Наличием водородных связей объясняется строение воды и льда и их свойства. Вода состоит из двух атомов водорода и одного атома кислорода. Молекула воды полярна, так как кислород обладает большей электроотрицательностью, чем водород, и оттягивает общие пары электронов на себя, приобретая частично отрицательный заряд. Атомы водорода, таким образом, оказываются частично заряженными положительно. Поэтому молекулы воды образуют меж собой водородные связи; при 370
С около 15% всех молекул воды соединены такими связями. Водородные связи образуют и другие молекулы. Молекулы фторида водорода, например, образуют зигзагообразные цепочки вследствие наличия водородной связи. За счѐт водородных связей формируется вторичная и третичная структура белка. Водородными связями между азотистыми основаниями, - пуринами и пиримидинами, - связаны меж собой две цепочки молекулы дезоксирибонуклеиновой кислоты, ДНК.
15.5. Комплементарность
Комплементарностью в химии вообще называют пространственное соответствие структур двух одинаковых или разных молекул, благодаря которому возможно образование между ними водородных связей
и осуществление межмолекулярного взаимодействия. В широком смысле под комплементарностью понимают также взаимное соответствие противоположных электростатических зарядов на молекулах и энергий сопряжѐнных реакций. Под сопряжѐнными реакциями понимают параллельно протекающие процессы, связанные друг с другом так, что стадия, сопровождающаяся выделением энергии, сопряжена со стадией, для осуществления которой необходимо потребление энергии. За счѐт структурной комплементарности осуществляется биохимическая связь по принципу «ключ – замок», или «замок-молния», и образуются такие сложные структуры, как четвертичная структура белков, а также вторичная и третичная структура нуклеиновых кислот. В узком смысле термин «комплементарность» применим к связи аденина с тимином
и гуанина с цитозином
(см. Лекцию 6), открытой Дж. Уотсоном и Ф. Криком в 1953 году и легшей в основу построенной ими модели двойной спирали ДНК. Этот тип комплементарности осуществляется вследствие образования водородных связей между протонодонорными и
протоноакцепторными группами в азотистых основаниях (см. Рис. 15.3)
Рис. 15.3. Водородные связи между комплементарными азотистыми основаниями, входящими в состав ДНК
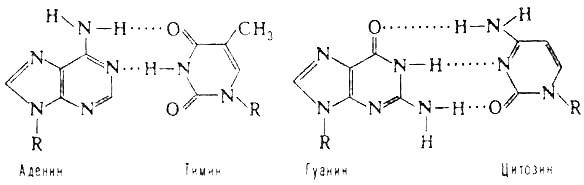
Именно это свойство ДНК делает возможным точное воспроизведение наследственной генной структуры. Модель Уотсона-Крика необыкновенно изящно решила проблему репликации гена[19]
. Если мы разведем в стороны две нити ДНК, а потом комплементарно нарастим на каждую из них по второй нити, то получим две абсолютно идентичные молекулы.
 Рис. 15.3. Репликация молекулы ДНК Рис. 15.3. Репликация молекулы ДНК
Хотя общий механизм репликации ДНК предельно прост, реальный биологический процесс является достаточно сложным. Во-первых, спиралевидная молекула должна «распрямиться» и одновременно образовать две новых молекулы. Процесс репликации протекает с огромной скоростью, нуклеотиды
соединяются в пары от 50 до 500 с секунду. Несмотря на такую высокую скорость, репликация происходит с невообразимой точностью: только один из миллиарда нуклеотидов в ДНК неправильно входит в пару. Для достижения такой точности и скорости в процесс вовлекаются десятки энзимов и белков, некоторые из них объединяются при этом в особые группы.
Несмотря на разнообразие видов химической связи, все они являются следствием электромагнитного фундаментального взаимодействия, то есть природа химической связи едина, а существующее различие между видами связи носит качественный характер.
15.6. Гибридизация
Часто в образовании нескольких химических связей участвуют электроны различных орбиталей одного и того же атома. Например, в молекуле метана четыре химические связи образованы перекрыванием одной s - и трех р - орбиталей атома углерода с четырьмя s - орбиталями атомов водорода. Следовало бы ожидать, что одна из связей в молекуле метана будет отлично от других, так как энергия и форма s - и р - орбиталей различна. Однако все четыре связи равноценны. Это объясняется тем, что при образовании молекул происходит гибридизация
- изменение энергии и формы атомных орбиталей. При этом образуются равноценные гибридные орбитали, причем гибридизация приводит к большому понижению энергии системы и повышению устойчивости молекулы. Тип гибридизации определяет пространственную структуру образовавшейся молекулы, что видно из следующей таблицы:
Таблица 15.2. Виды гибридизации и конфигурация молекул
| Молекулы
|
Кол-во эл-х пар
|
Гибридизация
|
Конфигурация
|
| CO2
, HgCl2
, BeF
|
2
|
Sp
|
Линейная
|
| BF3
, BCl3
|
3
|
sp2
|
Треугольник
|
| СH4
, SiH4
|
4
|
sp3
|
Тетраэдр
|
| SF6
|
6
|
d2
sp3
|
Октаэдр
|
Метод валентных связей не может объяснить некоторые существенные факты, например, парамагнитные свойства кислорода; упрочнения связи при отрыве электрона от некоторых молекул, например молекулы фтора; существование свободных радикалов. С помощью метода молекулярных орбиталей
(МО) можно объяснить факты, непонятные с точки зрения метода ВС.
15.7. Метод молекулярных орбиталей (ЛКАО МО)
Электроны в молекуле находятся на молекулярных орбиталях, охватывающих все ядра атомов. Эти молекулярные орбитали, захватывая весь объем атома, являются линейной комбинацией атомных орбиталей (ЛКАО). Каждый электрон в молекуле находится на определенном энергетическом уровне, обладает соответствующими квантовыми числами и подчиняется принципу Паули. Заполнение уровней электронами происходит в порядке возрастания энергии от низшего уровня к высшему. При сложении двух АО может возникнуть связывающая, несвязывающая и разрыхляющая
орбиталь. Вместо валентности в методе МО для характеристики связи используют такое понятие, как порядок связи,
или полуразность количества электронов на связывающих и разрыхляющих орбиталях.
АО могут взаимодействовать друг с другом по сигма-, пи- и дельта -типу. По сигма-типу комбинируются s-, pх
-, d- АО. По пи-типу комбинируются только py,z
- и d - АО. По дельта-типу комбинируются только некоторые d -АО орбитали. По уровню энергии молекулярные орбитали располагаются в следующем порядке в порядке возрастания энергии:
   1s < *1s< 2s < *2s < 2px
< 2py
= 2pz
< *2py
= *2pz
< *2px 1s < *1s< 2s < *2s < 2px
< 2py
= 2pz
< *2py
= *2pz
< *2px
Электронная структура молекулы водорода имеет вид:
H[1s1
] + H[1s1
]= H2
[( 1s)2
]
Диаграмма энергетических уровней молекулы водорода по ЛКАО МО приведена на Рис. 15.3.
Рис. 15.3. Диаграмма энергетических уровней молекулы водорода по методу
ЛКАО МО

Два электрона в молекуле Н2
занимают одну связывающую молекулярную орбиталь. Молекула Н2
стабильна, так как энергия связывающей орбитали меньше энергии орбиталей отдельных атомов. Порядок связи равен 2.
Для кислорода (два 1s-, два 2s- и четыре р-электрона) электронная структура молекулы следующая:
 О[1s1
2s2
2p4
]+О[1s1
2s2
2p4
]= О2
[KK( 2s)2
( *2s)2
( 2py
)2
( 2pz
)2
( 2px
)2
( *2py
)1
( *2pz
)1 О[1s1
2s2
2p4
]+О[1s1
2s2
2p4
]= О2
[KK( 2s)2
( *2s)2
( 2py
)2
( 2pz
)2
( 2px
)2
( *2py
)1
( *2pz
)1
 Буквами КК обозначаются заполненные уровни ( 1s)2
и ( *1s)2
.
В молекуле имеются два неспаренных электрона, что обуславливает ее парамагнитные свойства. На Рис. 15.4. диаграмма энергетических уровней молекулы кислорода по методу ЛКАО МО Буквами КК обозначаются заполненные уровни ( 1s)2
и ( *1s)2
.
В молекуле имеются два неспаренных электрона, что обуславливает ее парамагнитные свойства. На Рис. 15.4. диаграмма энергетических уровней молекулы кислорода по методу ЛКАО МО
Рис. 15.4. Диаграмма энергетических уровней молекулы кислорода по методу
ЛКАО МО
 В молекуле O2
два электрона с параллельными спинами оказались на двух В молекуле O2
два электрона с параллельными спинами оказались на двух 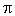 *-разрыхляющих молекулярных орбиталях с одинаковой энергией. Именно наличием этих неспаренных электронов и обусловлены парамагнитные свойства молекулы *-разрыхляющих молекулярных орбиталях с одинаковой энергией. Именно наличием этих неспаренных электронов и обусловлены парамагнитные свойства молекулы
кислорода, которые обнаруживаются, если охладить кислород до жидкого состояния.
Методы ВС и МО взаимно дополняют друг друга и объясняют свойства различных молекул. Они служат квантовомеханическим обоснованием теории химической связи А.М.Бутлерова, согласно которой свойства химических соединений определяются природой, количеством и взаимных расположением атомов.
15.8. Вопросы и задания
1. Как образуется химическая связь? Что такое валентность в современном понимании?
2. Что такое «электроотрицательность»?
3. Как кислотность оксидов связана со степенью окисления?
4. Укажите молекулу с самой прочной химической связью: H2
S, H2
Te, H2
O, H2
Se и тип связи.
5. В каком из приведенных соединений химическая связь будет больше всего близка к ковалентной? LiF; BeF2
; BF3
; CF4
6. Что такое сигма-, пи- и дельта-связь?
7. Что такое гибридизация? Какие типы гибридизации вы знаете, Какова связь между видом гибридизации и пространственным строением молекулы?
8. Опишите методом молекулярных орбиталей молекулы Н2,
N2
, O2
.
9. Определите заряд комплексного иона, степень окисления и координационное число комплексообразователя в соединениях:
[Cu(NH3
)4
]SO4
; К2
[HtCl6
]; K[Ag(CN)2
]. Напишите уравнения диссоциации этих соединений в водных растворах. Назовите эти соединения.
10. Как на основе принципа комплементарности объяснить репликацию молекулы ДНК?
Лекция 16. Химическая идентификация
Управление химическим производством, а также любая научноисследовательская работа по химии или химической технологии невозможны без химико-аналитического контроля
, то есть без знаний о качественном и количественном составе химических систем. Раздел химии, занимающийся химической идентификацией
, называется аналитической химией, или аналитикой
.
Аналитическая химия изучает методы исследования химического состава вещества или смеси веществ. В зависимости от того, требуется ли только обнаружить элемент или его соединение или же нужно определить его количественное содержание, различают качественный и количественный анализ
.
16.1. Качественный анализ
Обнаружение или, как говорят аналитики, открытие
элементов или ионов, входящих в состав исследуемого вещества, составляет предмет качественного анализа
. Открытие элементов или ионов осуществляется обычно путем перевода их в соединения, обладающие какими-либо характерными особенностями. Происходящее при этом химическое превращение называют аналитической реакцией
, а вещество, вызывающее такое превращение – реактивом, или реагентом
.
В зависимости от того, какими количествами вещества оперируют при выполнении аналитических реакций, различают макро-, микро- и полумикрометоды
качественного анализа. При макроанализе
исследуют сравнительно большие количества вещества, обычно около 1 г, при растворении вещества доводят объем раствора до 20-30 мл. Реакции выполняют в пробирках, поэтому часто этот метод называют «пробирочным анализом».
При микроанализе
имеют дело обычно с количествами вещества, примерно в 100 раз меньшими, то есть с несколькими миллиграммами твердого вещества или с несколькими десятками миллилитров раствора. При этом пользуются высокочувствительными реакциями, позволяющими обнаружить компоненты, присутствующим в анализируемом веществе в очень малых количествах. Реакции при микроанализе чаще всего выполняют либо микрокристаллоскопическим
, либо капельным
методом. При микрокристаллоскопическом методе реакции выполняют на предметном стекле и судят о присутствии элемента по характерной форме образующихся кристаллов, которые рассматривают под микроскопом. При капельном анализе на фильтровальную бумагу наносят в определенной последовательности капли исследуемого раствора и реактивов. В результате на бумаге получается окрашенное пятно, по цвету которого и судят о наличии в растворе открываемого элемента или иона. Полумикроанализ
занимает промежуточное положение между макро- и микроанализом. Количество исследуемого вещества в этом случае приблизительно равно 50 мг твердого вещества или 1 мл раствора.
Качественный анализ начинают с так называемых предварительных проб
. Для предварительных испытаний употребляют реакции, проводимые сухим путем
. При этом исследуемое вещество и соответствующие реактивы берут в твердом виде и нагревают их до высокой температуры. При нагревании анализируемого вещества из него могут выделяться газы, по запаху которых можно сделать предварительные выводы о составе анализируемой пробы.
Окраска пламени горелки при внесении в него анализируемого вещества (так называемые реакции окрашивания пламени
) указывает на наличие в пробе таких элементов, как литий (красный цвет пламени), натрий (желтый), калий (фиолетовый) или медь (зеленый). К реакциям сухим путем относятся также образование окрашенных перлов
(стекол) буры Na4
B4
O7
∙10H2
O или сплавление исследуемого твердого вещества с теми или иными плавнями, например, со смесями твердых карбонатов натрия и калия. К реакциям сухим путем относится и метод растирания исследуемого вещества с теми или иными твердыми реактивами. Так, например, можно обнаружить присутствие солей уксусной кислоты по запаху уксуса, выделяющегося при растирании небольшого количества соли в фарфоровой или агатовой ступке.
Главную же роль в качественном анализе играют реакции, выполняемые мокрым путем
, то есть реакции, происходящие между растворенными веществами. Для выполнения этих реакций вещество должно быть предварительно растворено в воде или в другом растворителе (например, в кислоте), если оно не растворяется в воде. В качественном анализе применяются только те реакции, которые сопровождаются внешним эффектом
, то есть характерным изменением свойств, которое легко обнаружить. К таким изменениям относятся:
 Выпадение (или растворение) осадка Выпадение (или растворение) осадка
 Изменение окраски раствора Изменение окраски раствора
Выделение газа
Внешний эффект, имеющий место при аналитической реакции, называется аналитическим сигналом
. Особенно часто применяются реакции, которые сопровождаются образованием осадков и изменением окраски растворов. При проведении реакций мокрым путем чаще всего пользуются растворами солей, кислот или растворимых гидроксидов. Как известно, все эти вещества являются электролитами, то есть в растворе диссоциируют на ионы. Ионы представляют собой свободные, раздельно существующие в растворе противоположно заряженные частицы (катионы и анионы), которые находятся в простой смеси друг с другом. Так, например, раствор смеси таких веществ, как HCl, KCl, NaCl, CaCl2
, FeCl3
является смесью катионов калий, натрия, кальция и железа (III) с хлорид-анионом. Таким образом, качественный анализ вещества или смеси веществ сводится к обнаружению (открытию) находящихся в растворе ионов. Количество наиболее важных катионов сравнительно невелико, всего около 25. Примерно столько же и наиболее известных анионов. Число же образуемых ими средних солей (не считая кислых и основных) составляет около 600. Поскольку все растворимые соли в растворе почти полностью диссоциируют на ионы, достаточно знать реакции всего лишь 50 ионов. Формулу индивидуального вещества часто можно установить уже при качественном испытании. Если, например, в исследуемом веществе были открыты только катион натрия и сульфат-анион, очевидно, что растворенное вещество представляет собой сульфат натрия. Благодаря электролитической диссоциации анализируемых веществ и реактивов химический анализ сильно облегчается. При анализе же недиссоциируюших соединений (в основном, органических) каждое вещество приходится распознавать по совокупности присущих только ему физических и химических свойств, что сильно затрудняет анализ.
Реакции аналитической химии являются, преимущественно, реакциями ионов. Каждая аналитическая реакция должна выполняться в строго определенных условиях. Эти условия зависят от свойств образующегося при реакции соединения; при несоблюдении этих условий результаты анализа окажутся недостоверными.
Наиболее важными характеристиками аналитической реакции являются ее чувствительность, специфичность и селективность
. При очень малых концентрациях определяемого вещества реакции не идут. Чем меньше концентрация вещества, при которой данная аналитическая реакция удается, тем выше чувствительность этой реакции. Например, реакция окрашивания пламени очень чувствительна, так как изменение окраски пламени происходит при очень малом количестве внесенного в пламя вещества.
Количественными характеристиками чувствительности служат открываемый минимум и предельное разбавление
. Открываемым минимумом называется наименьшее количество вещества или иона, которое может быть открыто посредством данной реакции при определенных условиях ее выполнения. Это количество обычно выражают в микрограммах (в миллионных долях грамма) и обозначают греческой буквой «гамма». Имеет значение не только абсолютное количество, но и концентрация определяемого иона в растворе. Поэтому наряду с открываемым минимумом указывают величину предельного разбавления, то есть наименьшую концентрацию вещества, при которой аналитическая реакция еще удается, как отношение 1:G, где G – весовое количество растворителя, приходящегося на 1 весовую часть открываемого вещества или иона (например, 1:1000 000).
Основная проблема качественного анализа состоит в том, что многие катионы и анионы дают одинаковые или сходные качественные реакции, то есть мешают определению
друг друга. Поэтому при проведении качественного анализа обязательно приходится проводить химическое разделение
ионов, пользуясь различной растворимостью их соединений и другими свойствами, которые химик-аналитик должен очень хорошо знать. В аналитической химии очень большое значение имеет опыт аналитика, а также особенная аккуратность и тщательность при проведении качественного определения. Не всегда просто узнать осадок или окраску в присутствии большого количества мешающих определению друг друга ионов; можно «потерять» небольшие количества присутствующего в анализируемой пробе вещества при разделении или «переоткрыть» отсутствующий в пробе ион, случайно загрязнив образец. Поэтому при применении аналитических реакций особое значение имеет специфичность
реакции. Специфической реакцией
на данный ион называют такую реакцию, которая позволяет открывать этот ион в присутствии любых других ионов. Например, при добавлении щелочи к раствору соли аммония выделяется аммиак, который легко узнать по резкому специфическому запаху. NH4
Cl + NaOH = NaCl + H2
O + NH3
Выделять аммиак в данных условиях могут только соли аммония, поэтому данная реакция является специфической для иона аммония NH4
+
.
Специфичные реакции весьма удобны, так как позволяют определять отдельные ионы, не считаясь с присутствием в растворе других ионов. При этом не имеет значения последовательность, в которой открываются отдельные ионы. Такое открытие ионов посредством специфических реакций в отдельных порциях раствора называется дробным анализом
. Однако, специфических реакций не так уж много. Чаще аналитику приходится иметь дело с реакциями, дающими одинаковый эффект с несколькими, а иногда и со многими ионами. Такие реакции называют избирательными, или селективными
. Чем меньше число ионов, реагирующих в данных условиях сходным образом, тем выше селективность данной реакции. Чаще всего прибегают к систематическому анализу
смеси ионов в растворе. При систематическом анализе сложной смеси прежде всего выделяют группы ионов, пользуясь селективными реакциями. Вещество, вызывающее такие реакции, называют групповым реагентом
. Групповой реагент позволяет отделить целую группу ионов, вызывающих специфическую реакцию, от групп других ионов. Сложная смесь распадается на ряд простых, анализ которых ведут при помощи специфических реакций на отдельные ионы. На основании различий в селективных реакциях все катионы делят на пять групп. В таблице 16.1. приведена классификация катионов в рамках классического сероводородного метода качественного анализа:
Таблица 16.1. Классификация катионов по сероводородному методу
| Группа
|
Характеристика группы
|
Состав группы
|
Групповой реагент
|
| 1
|
Сульфаты и карбонаты растворимы в воде
|
Катионы калия, натрия, аммония и др.
|
Группового реагента нет
|
| 2
|
Карбонаты нерастворимы в воде
|
Катионы кальция, стронция, бария, магния и др.
|
Групповой реагент
карбонат аммония
|
| 3
|
Сульфиды или гидроксиды (выпадающие при действии группового реагента из солей, подверженных гидролизу) растворимы в разбавленных кислотах
|
Катионы алюминия, хрома (III), железа
(II, III), марганца (II), цинка, никеля,
кобальта
|
| 4а
|
Хлориды
нерастворимы в воде,
сульфиды нерастворимы в разбавленных
кислотах
|
Катионы свинца (II), серебра (I), ртути (I) и др.
|
Групповой реагент – хлористоводородная кислота
|
| 4б
|
Сульфиды нерастворимы в разбавленных кислотах
|
Катионы меди (II), кадмия, висмута
(III), ртути (II) и др.
|
Групповой реагент – сероводород
|
| 5
|
Сульфиды растворимы в многосернистом аммонии и нерастворимы в разбавленных кислотах
|
Катионы мышьяка (III), олова (II), сурьмы (III) и др.
|
Групповой реагент дисульфид аммония
(NH4
)2
S2
|
Анионы делят на три группы:
Таблица 16. 2. Классификация анионов
| Группа
|
Характеристика группы
|
Состав группы
|
Групповой реагент
|
| 1
|
Соли бария не растворимы в воде
|
Сульфат, сульфит, тиосульфат, карбонат, ортофосфат, силикат, тетраборат, борат и др.
|
Хлорид бария в нейтральном или слабощелочном растворе
|
| 2
|
Соли серебра не растворимы ни в воде, ни в разбавленной азотной кислоте
|
Хлорид, бромид, иодид, сульфид и др.
|
Нитрат серебра в присутствии азотной кислоты
|
| 3
|
Соли бария и серебра растворимы в воде
|
Нитрат, нитрит, ацетат, др.
|
Группового реагента нет
|
Общая характеристика катионов первой группы
: первая группа катионов, как видно из таблицы 1, отличается от всех других групп тем, что соответствующие сульфиды, хлориды и карбонаты растворимы в воде. Первая группа не имеет собственного группового реагента, не осаждается групповыми реагентами других групп и при отделении их остается в растворе. В водных растворах все катионы этой группы бесцветны.
Некоторые специфические реакции катионов первой групп.
Катионы аммония
открывают, приливая к раствору соли (около 1 мл) 2-3 мл раствора едкого натра или едкого кали. Нагревают раствор в пробирке до кипения. Осторожно нюхают выделяющиеся из пробирки газы и по запаху определяют наличие аммиака, выделяющегося в результате реакции:
NH4
+
+ OH-
= NH4
OH → NH3
+ H2
O
Ионы аммония мешают определения катионов натрия и калия, поэтому их необходимо удалить из раствора. Для этого, перелив раствор в фарфоровую чашечку, выпаривают его под вентиляцией досуха и прокаливают до прекращения выделения белого дыма. После полного удаления ионов аммония, оставляют часть сухого остатка для пробы на пламя при определении катионов натрия или калия. Остаток растворяют в небольшом количестве воды для обнаружения в нем других ионов.
Катионы калия и натрия
проще всего открываются в реакциях окрашивания пламени. Металлическую (лучше всего платиновую) проволочку, конец которой загнут в ушко, вносят в пламя горелки, чтобы убедиться, что проволочка чиста и сама не окрашивает пламя. Если окрашивание наблюдается, необходимо промыть проволочку раствором соляной кислоты и прокалить до исчезновения окрашивания. Затем раскаленной проволочкой прикасаются к мелкому порошку соли калия
и приставшие к проволочке крупинки вносят в пламя, наблюдая возникновение бледно-фиолетового
окрашивания последнего. При использовании раствора соли калия реакция удается хуже. Соли натрия
окрашивают пламя в ярко-желтый
цвет.
Общая характеристика катионов второй группы
: наиболее важное для анализа свойство катионов второй группы состоит в образовании ими нерастворимых карбонатов при действии группового реагента углекислого аммония. Использование карбонатов калия или натрия в качестве группового реагента привело бы к введению в анализируемый раствор ионов калия и натрия и искажению результатов анализа. В то же время, сульфиды катионов второй группы растворимы в воде, что позволяет отделять их от катионов третьей, четвертой и пятой групп. Все металлы, образующие катионы второй группы, относятся ко второй группе периодической системы элементов Д.И.
Менделеева и являются двухвалентными. Осаждение катионов второй группы ведут в присутствии гидроксида аммония и небольшого количества хлорида аммония при умеренном нагревании раствора.
Некоторые специфические реакции катионов второй группы
. Реакции катиона бария
. Серная кислота и ее растворимые соли (содержащие сульфат-анион) образуют с катионом бария белый осадок сульфата бария:
Ва2+
+ SO4
2-
= Ba SO4
Хромовокислый калий (содержащий в растворе хромат-ион) образует с катионом бария желтый осадок хромовокислого бария: Ва2+
+ CrO4
2-
= Ba SO4
Реакции катиона кальция
: соли серной кислоты вызывают выпадение белого осадка сульфата кальция из сравнительно концентрированных растворов его солей. Осадок сернокислого кальция растворяется в присутствии сернокислого аммония (вследствие образования комплексной соли), поэтому, добавляя сернокислый аммоний, можно осадить катионы бария и отделить их от катионов кальция, которые останутся в растворе.
Ca2+
+ SO4
2-
= CaSO4
Общая характеристика третьей группы катионов
: основное отличие катионов этой группы от других состоит в том, что их сульфиды не растворимы в воде, но растворяются в разбавленных кислотах или разлагаются водой вследствие гидролиза с образованием растворимых в кислотах гидроксидов. Групповой реагент сульфид аммония осаждает катионы железа, марганца, цинка, никеля и кобальта в виде сульфидов:
FeSO4
+ (NH4
)2
S = FeS + (NH4
)2
SO4
2 FeCl3
+ (NH4
)2
S = Fe2
S3
+ 6NH4
Cl MnSO4
+ (NH4
)2
S = MnS + (NH4
)2
SO4
Сульфиды железа, никеля и кобальта черного цвета, сульфид марганца телесного цвета, а сульфид цинка – белого цвета. Все они, кроме сульфидов никеля и кобальта, растворимы в разбавленной соляной кислоте:
ZnS + HCl = ZnCl2
+ H2
S; Fe2
S3
+ 6HCl = 2FeCl3
+3Н2
S
При растворении сульфида железа (III) в соляной кислоте образуется хлорид двухвалентного железа, так как происходит реакция окислениявосстановления между окислителем трихлоридом железа и восстановителем сероводородом.
Fe2
S3
+ 4HCl = 2FeCl2
+ S + 2 H2
S
Катионы алюминия и хрома осаждают в виде гидроксидов, потому что последние менее растворимы, чем соответствующие сульфиды. 2AlCl3
+ 3(NH4
)2
S + 6H2
O = 2Al(OH)3
+ 6NH4
Cl + 3H2
;
Катионы третьей группы осаждают групповым реагентом из растворов, нейтрализованных гидроксидом аммония, для исключения растворения выпадающих в осадок сульфидов в кислоте и для подавления гидролиза солей. Сульфиды катионов третьей группы часто образуют коллоидные растворы, поэтому их осаждение ведется из нагретого до кипения раствора с добавлением коагулятора хлорида аммония.
Некоторые специфические реакции катионов третьей группы
. Железо
образует два ряда солей с катионом Fe2+
и с катионом Fe3+
. Реакции обоих катионов различны. Роданид калия KCNS
(или аммония NH4
CNS
) с ионом Fe3+
роданид железа кроваво-красного цвета:
FeCl3
+ KCNS = Fe(CNS)3
+ 3 KCl
Гексацианоферрат (II) калия дает с ионом Fe3+
тѐмно-синий осадок, называемый берлинской лазурью:
4FeCl3
+ 3K4
[Fe(CN)6
] = Fe4
[Fe(CN)6
]3
+ 12KCl
Гексацианоферрат (III) калия дает с ионом Fe2+
тѐмно-синий осадок турнбулевой сини (иного оттенка, чем берлинская лазурь):
3FeSO4
+ 2K3
[Fe(CN)6
] = Fe3
[Fe(CN)6
]2
+ 3K2
SO4
Катион алюминия
осаждается гидроксидом калия или натрия с образованием белого аморфного осадка гидроксида алюминия, который растворяется в кислотах и щелочах:
AlCl3
+ 3NaOH = Al(OH)3
+ 3NaCl; Al(OH)3
+ NaOH = NaAlO2
+ H2
O
Общая характеристика катионов четвертой группы
. При действии группового реагента сероводорода (который обычно получают в лаборатории в аппаратах Киппа действием хлористоводородной кислоты на сернистое железо) на катионы четвертой группы выпадают осадки соответствующих сульфидов. Осаждение ведут в кислой среде, чтобы отделить катионы четвертой группы от катионов третьей группы, сульфиды которых растворяются в разбавленных кислотах. Сульфиды серебра, свинца, ртути (II), меди черного цвета; из растворов солей кадмия выпадает осадок ярко-желтого цвета, это специфическая реакция катиона кадмия:
CdSO4
+ H2
S = СdS + H2
SO4
Некоторые специфические реакции катионов четвертой группы.
Катион серебра
определяют, осаждая его в виде хлорида, нерастворимого в кислотах:
AgNO3
+ HCl = AgCl + HNO3
Специфической реакцией катиона свинца
является образование характерного ярко-желтого йодида свинца:
Pb(NO3
)2
+ KJ = PbI2
+ KNO3
Катион меди Cu2+
имеет в растворе синий цвет. Едкие щелочи NaOH
и
KOH
осаждают его в виде голубого осадка гидроксида меди, который чернеет при нагревании вследствие превращения в оксид меди CuO. При добавлении избытка аммиака к раствору соли меди образуется сульфат тетрамминмеди ярко-синего цвета:
CuSO4
+ 4NH3
= [Cu(NH3
)4
]SO4
Анализ пятой группы катионов представляет известные трудности и в данном пособии не рассматривается.
Систематический ход анализа
катионов начинают с открытия ионов аммония и железа в отдельных порциях раствора. Затем последовательно отделяют катионы четвертой, третьей и второй групп путем прибавления соответствующего группового реагента в надлежащих условиях. Осадки и фильтраты анализируют отдельно, тщательно производя разделение катионов и открывая их при помощи специфических реакций. В ходе анализа катионов обычно можно сделать вывод о присутствии в анализируемом растворе тех или иных анионов. Поэтому при открытии анионов сравнительно редко прибегают к реакциям разделения анионов групповыми реагентами и для их открытия используют специфические реакции.
Специфические реакции некоторых анионов
: сульфат-ион образует с ионом бария нерастворимый в кислотах осадок сульфата бария. Сульфит-тон определяют по характерному запаху сернистого газа при подкислении раствора и по способности обесцвечивать раствор перманганата калия. Тиосульфат-ион определяют по помутнению раствора при подкислении:
Na2
S2
O3
+ 2 HCl = 2 NaCl + H2
S2
O3
H2
S2
O3
= H2
O + S + SO2
Карбонат-ион
дает с хлористым барием белый осадок, растворимый в кислотах. Сильные кислоты разлагают все углекислые соли с выделением диоксида углерода. Хлорид-ион
образует с азотнокислым серебром белый осадок, не растворимый в азотной кислоте, но растворяющийся при действии раствора аммиака. Йодидион
в таких же условиях образует осадок желтого цвета, не растворимый в аммиаке. Добавление растворимой соли свинца приводит к выпадению осадка йодистого свинца ярко-желтого цвета.
Общий ход анализа какого-либо вещества состоит из следующих этапов:
1) Подготовка вещества к анализу: твердое вещество тщательно измельчают и растирают в ступке до мелкодисперсного состояния. Металлы и металлические сплавы превращают в стружку или в опилки. Раствор с осадком фильтруют и анализируют осадок и фильтрат раздельно.
2) Подготовленное к анализу вещество делят на три части: одну оставляют в запасе, другую употребляют для открытия катионов, а третью – для открытия анионов.
3) Перевод вещества в раствор при помощи подходящего растворителя. Подавляющее большинство веществ растворяется в воде или в разбавленной соляной кислоте. Труднорастворимые вещества переводят в раствор при помощи концентрированной азотной кислоты или «царской водки» - смеси 3 частей соляной и 1 части азотной концентрированных кислот. Прибегают также к сплавлению вещества с карбонатами калия или натрия.
4) Открытие катионов.
5) Открытие анионов.
16.2. Количественный анализ
При количественном определении необходимо ответить на вопрос о точном количестве
того или иного вещества, содержащегося в данном образце. К химическим (
или так называемым классическим) методам
количественного анализа относятся весовой
(гравиметрический) и объемный
(титриметрический) методы. При определении неизвестного количества данного элемента весовым способом необходимо получить соответствующий реакцией осаждения наименее растворимое его соединение, отфильтровать осадок, высушить его до постоянного веса и взвесить с необходимой точностью. Затем необходимо рассчитать содержание данного элемента в исходной пробе в виде того соединения, в котором данный элемент присутствует в анализируемой пробе.
Например, если мы хотим определить содержание хлористого бария в пробе, где он может быть загрязнен примесями других солей, нам следует растворить точно взвешенное количество пробы (навеску) в воде. Затем необходимо осадить барий в виде сульфата (а не карбоната, например, так как из всех нерастворимых соединений бария сульфат наименее растворим, и мы извлечем из раствора максимальное его количество), отфильтровать осадок, высушить его и взвесить на аналитических весах. Далее следует пересчитать сульфат бария на хлорид, используя следующее соотношение:
m
BaCl
2
 BaSO
4
, (16.1) BaSO
4
, (16.1)
где F- фактор пересчѐта сульфата бария на хлорид бария, определяемый отношением молярных масс соответствующих солей:
M
BaCl
2
 F
(16.2) F
(16.2)
M
BaSO
4
Весовой метод определения очень простой и дешевый, но к его недостаткам относятся следующие:
1. Не все элементы имеют нерастворимые соединения, которые могут использоваться в качестве весовой формы;
2. многие элементы соосаждаются в одинаковых условиях и мешают определению друг друга;
3. Весовой метод требует больших затрат времени;
4. Малая чувствительность анализа (около 0,01%)
Объемный анализ
основан на соотношении, вытекающем из закона эквивалентов:
 N
1 V
1 N
2 V
2 (16.3) N
1 V
1 N
2 V
2 (16.3)
где N1
и N2
– нормальные концентрации[20]
, а V1
и V2
– объемы реагирующих растворов.
В объѐмном методе анализа к известному объему известного вещества неизвестной концентрации добавляют из бюретки по каплям известное вещество известной концентрации до тех пор, пока индикатор не покажет, что реакция закончена. Этот процесс называется титрованием (от французского слова titre
- содержание). Так как в соответствии с вышеприведенным равенством (16.3) вещества реагируют всегда в эквивалентных отношениях, можно определить нормальную концентрацию определяемого вещества.
N
1
V
1
N
2
(16.4)
V
2
Существует множество способов объемного анализа, в которых используются реакции различных типов: нейтрализации, осаждения, комплексообразования, окисления-восстановления и др.
Рассмотрим простейший случай, когда необходимо определить неизвестную концентрацию гидроксида натрия в растворе. Будем исследовать нашу пробу при помощи реакции нейтрализации – реакции гидроксида натрия с кислотой. В основе реакции нейтрализации лежит взаимодействие водородных ионов с ионами гидроксила с образованием воды:
H+
+ OH-
= H2
O
 Так как мы определяем содержание щелочи, то наш метод будет называться алкалиметрией
(alkali
означает щелочь); если бы нам нужно было определить содержание кислоты, мы бы воспользовались ацидиметрией
(acid
значит кислота). Будем добавлять к точно отмеренному объему щелочи с неизвестной концентраций по каплям соляную кислоту с концентрацией 0,1N (если концентрация кислоты не равна точно 1,000 моль-эквиваленту в литре, нужно определить ее поправочный коэффициент К). При титровании сильной щелочи сильной кислотой нейтрализация наступает, когда рН раствора равен 4 –5. При этом рН индикатор метиловый оранжевый меняет оранжевый цвет на красный, а индикатор фенолфталеин, окрашенный в малиновый цвет в щелочном растворе, обесцвечивается. Отберем мерной пипеткой три пробы анализируемого раствора гидроксида натрия по 5,0 мл в конические колбы, добавим по 3-4 капли индикатора и будем по капле добавлять к раствору в колбе кислоту из бюретки, постоянно перемешивая содержимое колбы. Под колбу рекомендуется подложить лист белой бумаги для того, чтобы точно заметить момент изменения окраски индикатора. В этот момент зафиксируем объем добавленной кислоты по показаниям бюретки, а затем добавим еще 2-3 капли кислоты, чтобы посмотреть, стала ли окраска раствора интенсивнее. Если это так, то прибавим к отмеченному расходу кислоты объем добавленных капель. Повторим опыт еще два раза, рассчитаем среднее значение объема израсходованной на титрование кислоты и вычислим нормальность раствора щелочи. Допустим, что на титрование 5 мл раствора щелочи израсходовано 10,0 мл кислоты. Тогда нормальность раствора щелочи равна: 0,110,0/5,0 = 0,2N. Это означает, что в 1 литре исследуемого раствора содержится 0,2 моль-эквивалента NaOH
, или 40 Так как мы определяем содержание щелочи, то наш метод будет называться алкалиметрией
(alkali
означает щелочь); если бы нам нужно было определить содержание кислоты, мы бы воспользовались ацидиметрией
(acid
значит кислота). Будем добавлять к точно отмеренному объему щелочи с неизвестной концентраций по каплям соляную кислоту с концентрацией 0,1N (если концентрация кислоты не равна точно 1,000 моль-эквиваленту в литре, нужно определить ее поправочный коэффициент К). При титровании сильной щелочи сильной кислотой нейтрализация наступает, когда рН раствора равен 4 –5. При этом рН индикатор метиловый оранжевый меняет оранжевый цвет на красный, а индикатор фенолфталеин, окрашенный в малиновый цвет в щелочном растворе, обесцвечивается. Отберем мерной пипеткой три пробы анализируемого раствора гидроксида натрия по 5,0 мл в конические колбы, добавим по 3-4 капли индикатора и будем по капле добавлять к раствору в колбе кислоту из бюретки, постоянно перемешивая содержимое колбы. Под колбу рекомендуется подложить лист белой бумаги для того, чтобы точно заметить момент изменения окраски индикатора. В этот момент зафиксируем объем добавленной кислоты по показаниям бюретки, а затем добавим еще 2-3 капли кислоты, чтобы посмотреть, стала ли окраска раствора интенсивнее. Если это так, то прибавим к отмеченному расходу кислоты объем добавленных капель. Повторим опыт еще два раза, рассчитаем среднее значение объема израсходованной на титрование кислоты и вычислим нормальность раствора щелочи. Допустим, что на титрование 5 мл раствора щелочи израсходовано 10,0 мл кислоты. Тогда нормальность раствора щелочи равна: 0,110,0/5,0 = 0,2N. Это означает, что в 1 литре исследуемого раствора содержится 0,2 моль-эквивалента NaOH
, или 40 0,2 = 8г/л NaOH
. 0,2 = 8г/л NaOH
.
Титрование значительно сокращает время определения по сравнению с весовым методом, этот процесс можно автоматизировать, заменив визуальное наблюдение за окраской индикатора электрохимическими методами, однако чувствительность
объемного метода составляет всего 0,1%.
Развитие новой техники, в том числе радиоэлектроники, атомной энергетики, лазерной техники связано с применением веществ высокой чистоты. Самые незначительные примеси порядка 10-5
– 10-8
% делают материалы непригодными для использования их в новых технологиях. Бурно развивающаяся промышленность потребовала новых методов анализа, так как классические весовой и объемный методы вследствие малой чувствительности оказались совершенно непригодными для определения малых количеств примесей. Возникшая проблема была разрешена широким использованием разнообразных физических и физико-химических методов анализа
.
16.3. Физические и физико-химические методы анализа
Определить состав разнообразных веществ можно, не прибегая к химическим реакциям. Физические методы анализа
основаны на изучении таких физических свойств исследуемого вещества, как его спектры поглощения или испускания, электропроводности, потенциала электрода, опущенного в исследуемый раствор, показателя преломления света, флуоресценции, радиоактивности и т.п. Если для анализа состава вещества используются химические реакции, протекание которых сопровождается изменением физических свойств анализируемой системы, например, ее цвета, флуоресценции, электропроводности и пр., то такие методы анализа принято называть физико-химическими
.
В аналитической практике, как правило, не разделяют физические и физико-химические методы анализа и объединяют их под общим называнием физико-химические методы анализа
. По сравнению с классическими методами, физико-химические методы являются гораздо более чувствительными, то есть помогают определять очень малые количества вещества (до 10-9
%). Однако по точности эти методы уступают классическим методам анализа. Точность весового и объемного метода достигает сотых долей процента, тогда как ошибка определения физико-химическими методами составляет 5-10%, а иногда и больше. Из этого вытекает важный вывод, что не следует прибегать к особо чувствительным методам анализа, когда в этом нет настоятельной необходимости. Ни один из аналитических методов не является универсальным. Выбирать метод анализа необходимо исходя из характера исследуемого объекта, его назначения, агрегатного состояния, концентрации, наличия примесей, а также учитывая цели анализа, требуемую точность, срок исполнения анализа и т.д. Химиканалитик может успешно разрешить любую стоящую перед ним задачу, только если он владеет самыми разнообразными методами анализа и умеет правильно их сочетать.
Не следует путать аналитические физико-химические методы анализа
и физико-химический, или термический анализ
, позволяющий изучать физические свойства систем в зависимости от их химического состава посредством фазовых диаграмм, или диаграмм плавкости (см. Лекцию 9). Термический анализ широко применяется для изучения свойств различных металлических сплавов и в данной главе не рассматривается.
К химическим методам анализа относятся весовой, объемный и газовый анализ. Физико-химические методы анализа делятся на следующие основные группы: электрохимические; спектральные (оптические); хроматографические; масс-спектрометрические
. В основе всех этих методов анализа лежит количественная зависимость между концентрацией определяемого вещества (компонента) и аналитическим сигналом – измеряемой в данном методе физической характеристикой вещества.
К основным электрохимическим методам анализа относятся потенциометрический, кондуктометрический
и основанные на законе Фарадея электровесовой, амперометрический, полярографический и кулонометрический
методы анализа. Аналитическим сигналом в электрохимических методах анализа является изменение таких характеристик, как стандартный электродный потенциал; количество электричества, прошедшего через раствор; электропроводность раствора, сила тока в зависимости от концентрации анализируемого раствора или изменения ее в результате химических или электрохимических реакций.
Прямым потенциометрическим методом
измеряют концентрацию различных ионов в растворе (например, Н+
, NH4
+
, NO3
-
, CO3
2-
).
Аналитическим сигналом служит изменение потенциала так называемого селективного электрода, который реагирует только на определенный находящийся в растворе ион. Зависимость аналитического сигнала от концентрации определяемого иона выражается уравнением Нернста:
 lgC
(16.5) n lgC
(16.5) n
где  - электродный потенциал системы, а С – концентрация определяемого иона. - электродный потенциал системы, а С – концентрация определяемого иона.
Самым распространенным прибором для ионометрии является рН-метр
(см. Рис. 14.1).
К группе спектральных (оптических) методов
относятся следующие методы:
 Эмиссионный спектральный анализ
– физический метод, основанный на изучении спектров излучения паров анализируемого вещества. Анализируемое вещество помещают в дуговой или искровой разряд, где оно испаряется и излучает свет с характерными длинами волн. Аналитическим сигналом здесь является интенсивность (яркость) определенной спектральной линии. Эмиссионный спектральный анализ
– физический метод, основанный на изучении спектров излучения паров анализируемого вещества. Анализируемое вещество помещают в дуговой или искровой разряд, где оно испаряется и излучает свет с характерными длинами волн. Аналитическим сигналом здесь является интенсивность (яркость) определенной спектральной линии.
Атомно-абсорбционная спектроскопия
основана на способности свободных атомов металла, которые образуются в восстановительном пламени, поглощать световую энергию при характерных для каждого элемента длинах волн. Этот метод используется в последнее время очень широко. Аналитическим сигналом является снижение интенсивности света той длины волны, которая поглощается анализируемым веществом.
Метод молекулярной абсорбционной спектроскопии
основан на поглощении окрашенным раствором света определенной длины волны.
Фотометрические методы
основаны на поглощении видимого света. Так, например, раствор, окрашенный в красный цвет, поглощает зеленый свет тем больше, чем интенсивнее окраска раствора (чем выше концентрация определяемого вещества). Поглощение света раствором (оптическая плотность D
) связана с концентрацией раствора законом Бугера-Ламберта-Бэра:
 DCl
(16.6) DCl
(16.6)
где ε – постоянный для данного соединения молярный коэффициент поглощения, l – толщина поглощающего слоя, С – молярная концентрация определяемого вещества.
Хроматографические методы анализа
применяются для разделения сложных смесей газов и жидкостей. Эти методы основаны на различном поглощении отдельных компонентов анализируемой смеси различными поглотителями. Здесь аналитическим сигналом является время выхода данного компонента из хроматографической колонки и величина «пика» определяемого вещества. Например, при анализе смеси газов, содержащей кислород, азот, диоксид азота и оксид углерода, смесь пропускают при низкой температуре через силикагель, служащий поглотителем (адсорбентом
). Поскольку силикагель адсорбирует разные газы с разной интенсивностью, при нагревании поглотителя происходит десорбция сначала кислорода (через 3 мин), затем (через 4 мин) азота, диоксида азота (через 10 мин) а затем оксида углерода (через 13 мин). Самописец фиксирует выход газов и их количественное содержание в виде пиков на хроматограмме на основе измерения теплопроводности, плотности, теплоты сгорания или диэлектрической проницаемости газов при помощи соответствующего детектора
. Подбирая соответствующие поглотители и элюенты («вымыватели» поглощенных веществ), а также соответствующее аппаратное оформление, можно разделять и анализировать сложные смеси неорганических и органических веществ. Наиболее перспективным современным хроматографическим методом является высокоэффективная жидкостная хроматография (ВЭЖХ).
Масс-спектрометрические методы анализа
основаны на определении массы отдельных атомов и молекул в результате комбинированного воздействия на них электрического и магнитного полей. В последнее время получило широкое распространение объединение хроматографических и масс-спектрометрических методов анализа. Хромато-масс-спектрометры
позволяют производить количественный и качественный анализ сложных соединений и смесей различных веществ. Существуют также методы анализа, основанные на измерении ядерных характеристик атомов, например, метод ядерно-магнитного резонанса ЯМР и метод ядернопарамагнитного резонанса ЯПР.
16.4. Вопросы и задания:
1. Что такое аналитический сигнал?
2. Опишите специфические реакции наиболее распространенных катионов и анионов.
3. Какова масса осадка, образующегося при смешении 0,1М раствора хлорного железа FeCl3
и 150 мл 0,2 М раствора гидроксида натрия NaOH?
4. Какой объѐм 0,1N раствора гидроксида калия КОН потребуется для нейтрализации 20 мл 0,15 мл раствора серной кислоты H2
SO4
?
5. Определите концентрацию раствора едкого кали КОН, если на нейтрализацию 0,035л фосфорной кислоты Н3
РО4
с концентрацией 0,3N израсходовано 0,02 л раствора КОН.
6. Перечислите методы физико-химического анализа с указанием аналитического сигнала, зависящего от содержания определяемого элемента.
7. Какой метод анализа был использован для определения хрома? Что является в данном случае аналитическим сигналом?
А) Перенесите 50 мл отфильтрованной пробы в колбу емкостью 125 мл.
Б) Добавьте к пробе 2,0 мл раствора дифенилкарбазида и перемешайте раствор.
В) Немедленно добавьте к каждому раствору 5,0 мл раствора фосфорной кислоты (1:1) и перемешайте растворы.
Г) Выдержите растворы 15 минут для развития окраски. Измерьте их светопоглощение в течение 30 минут после добавления раствора дифенилкарбазида на длине волны 540 нм в кювете длиной не менее 10 мм.
Д) Определите концентрацию хрома в пробе (Сr +6
) в мг/л по градуировочному графику (см. 12.3)
9. Какой метод анализа используется для определения меди? Что является в данном случае аналитическим сигналом?
А) Отмерьте 100, 0 мл хорошо перемешанной подкисленной пробы в стакан или колбу на 125 мл.
Б) Добавьте к каждой пробе по 5 мл HCl (пл. 1,19 г/см3
).
В) Распылите каждую обработанную пробу в пламя и определите ее абсорбцию и концентрацию на длине волны 324,7 нм. Между определениями распыляйте в пламя азотную кислоту HNO3
(1:499).
10. Каким методом можно разделить пигменты, из которых состоит хлорофилл зеленых листьев, и определить их количественный состав?
а) потенциометрическим; б) эмиссионным спектральным; б) методом атомноабсорбционной спектроскопии; г) хроматографическим методом; д) фотометрическим методом.
Лекция 17. Основы биохимических процессов и их применение в технологической отрасли
Биохимия
изучает химические процессы, происходящие в живых организмах. Все живые организмы, от простейших бактерий до высших животных, являются самыми высокоорганизованными системами
химических соединений. Можно назвать эти системы биологическим уровнем
организации вещества.
17.1. Клетка – основа жизни
Все живые организмы состоят из клеток. Клеткой называется простейшая биологическая система, способная жить. Основным отличием живых систем от неживых является способность живых организмов воспроизводиться
на основе генетического кода
и матричного синтеза
. Ещѐ одной важнейшей функцией живых систем является постоянный обмен веществ
с окружающей средой.
Существует множество одноклеточных живых организмов. Более сложные организмы, в том числе растения и животные, многоклеточные, их тела состоят из множества клеток различных видов, которые не могут существовать отдельно от организма. Все, что происходит в организме, происходит на клеточном уровне. Будучи открытыми системами
, клетки постоянно обмениваются энергией и веществом с окружающей средой.
Любой современный организм состоит из одного из двух типов клеток: прокариотов и эукариотов.
Прокариоты
- это клетки без ядра
.
Из них состоят бактерии и так называемые сине-зеленые водоросли. Все клетки растений и животных являются эукариотами
. Внутри клетки имеются различные образования, которые называются органеллами
(маленькими органами). Самой важной органеллой у эукариотов является ядро
. Ядро содержит генетический материал, молекулу ДНК
,
состоящую из определенного количества хромосом
для каждого вида клеток-эукариотов. Ядро имеет оболочку,
представляющую собой двойную мембрану
. Все пространство клетки ограничивает плазменная мембрана
.
Между оболочками ядра и клетки находится цитоплазма. Большинство реакций метаболизма протекает в цитоплазме. В цитоплазме, в среде полужидкого цитозола
,
располагаются различные органеллы.
Цитоплазму заполняет эндоплазматическая сетка (ЭС), целый лабиринт из мембран, образующий мешочки и трубочки, которые отделяют содержимое сетки от цитоплазмы. Разнообразные белки производятся рибосомами
, прикрепленными к мембранам ЭС, которые играют также основную роль в сборке других клеточных мембран. Органеллой, которая превращает энергию в такие формы, которые могут быть использованы клеткой, является митохондрия
. Ещѐ одна разновидность мембранной органеллы в цитоплазме, механизм, или аппарат Гольджи, состоит из нагромождения плоских мешочков, которые также играют важную роль в синтезе, очистке, накоплении, сортировке клеткой химических продуктов. Другими органеллами клетки являются лизосомы
,
содержащие смеси энзимов, которые ускоряют гидролиз макромолекул; микротела
, т.е. разнообразные группы определенных энзимов, осуществляющих процессы метаболизма, и др.
В отличие от клеток животных, клетки растений содержат органеллы, которые называются пластидами. Одним из важнейших пластидов является хлоропласт
,
содержащий хлорофилл,
который осуществляет фотосинтез
, превращая солнечный свет в химическую энергию, запасаемую в сахарах и других органических молекулах. Другой важной органеллой в клетках растений является большая центральная вакуоля
,
в которой запасаются химические вещества. Она также функционирует как лизосома и, увеличиваясь в размерах, играет важную роль в процессе роста растений. С наружной стороны плазменной мембраны у растений имеется толстая клеточная стенка
, которая определяет форму клетки и защищает ее от повреждений. Но
Рис. 17.1 схематически изображены основные органеллы клетки.
Рис. 17.1. Основные органеллы клетки
 Живая клетка состоит на 70 – 95 % из воды, всѐ остальное – это органические вещества. Основными классами органических соединений, входящих в состав клетки, являются углеводы
(полисахариды и моносахариды), липиды
(жиры, стероиды), белки
(протеины) и нуклеиновые кислоты
. Белок составляет Живая клетка состоит на 70 – 95 % из воды, всѐ остальное – это органические вещества. Основными классами органических соединений, входящих в состав клетки, являются углеводы
(полисахариды и моносахариды), липиды
(жиры, стероиды), белки
(протеины) и нуклеиновые кислоты
. Белок составляет
более 50% сухого остатка любой клетки и участвует во всех происходящих в ней процессах.
Углеводы, или сахара, делятся на моносахариды и полисахарида. Важнейшими для живых систем моносахаридами являются гексозы, в первую очередь, глюкоза
С6
Н12
О6
, а также пентозы рибоза
С5
Н10
О5
и дезоксирибоза
С5
Н10
О4
. Полисахариды можно представить как полимерные молекулы, состоящие из остатков молекул моносахарида глюкозы С6
Н12
О6
. Сахариды синтезируются зелѐными растениями в процессе фотосинтеза, который можно представить в виде упрощѐнной схемы образования глюкозы:
 6CО
2 6CО
2
 С
6
H
12
O
6
6O
2
; H
2815,8 кДж
(17.1) хлорофилл моль С
6
H
12
O
6
6O
2
; H
2815,8 кДж
(17.1) хлорофилл моль
Где h
 - кванты солнечной энергии, а хлорофилл – составная часть хлоропласта растений - катализатор, ускоряющий процесс фотосинтеза. Глюкоза является основным источником энергии для всех живых систем, то есть углеводы выполняют в жизнедеятельности организмов энергетическую функцию
. - кванты солнечной энергии, а хлорофилл – составная часть хлоропласта растений - катализатор, ускоряющий процесс фотосинтеза. Глюкоза является основным источником энергии для всех живых систем, то есть углеводы выполняют в жизнедеятельности организмов энергетическую функцию
.
Жиры, или липиды
, представляют собой сложные эфиры трѐхатомного спирта глицерина и насыщенных (жирных) карбоновых кислот с 16-28 атомами в углеродной цепи. В состав молекулы жира могут входить одинаковые или разные кислотные остатки. Примерами таких жиров являются тристеарин (1) и пальмитоолеостеарин (2).
1) СН2
-ОС(О)-С17
H35
СН2
-ОС(О)-С17
Н35
СН
2-ОС(О)-С
17Н
35
2) СН2
-О-С(О)-С15
Н31
СН2
-О-С(О)-С17
Н33
СН2
-О-С(О)-С17
Н35
Жиры являются хорошим источником энергии, так как полностью «сгорают» в организме, выделяя при этом вдвое больше энергии, чем такое же количество углеводов или белков.
Строение белков и нуклеиновых кислот как природных полимеров было рассмотрено в Лекции 6.
17.2. Метаболизм
Живая клетка – это целое химическое производство, где на микроскопическом пространстве происходят тысячи химических реакций. Метаболизмом
называется возникающее при этом свойство живой материи управлять этим множеством взаимодействий при обмене веществ с окружающей средой. Слово метаболизм
происходит от греческого слова, metabolus
означающего изменение
, поэтому его часто путают с обменом веществ
. Однако метаболизм не обмен веществ, а механизм управления
этим обменом. Метаболизм можно представить в виде тщательно продуманной «дорожной карты» десятков тысяч реакций, происходящих в клетке, сходной с картой интенсивных транспортных потоков большого города. Реакции проходят по замысловатым разветвленным «метаболическим тропинкам», на которых происходит постепенное превращение молекул. На некоторых «метаболических дорожках» энергия высвобождается при разложении сложных соединений на простые составляющие. Эти дорожки называются катаболическими.
Главные «магистрали» здесь – это процессы клеточного дыхания,
в которых глюкоза окисляется до двуокиси углерода и воды:
 С6
Н12
О6
→ 6СО2
+ Н2
О; H
2815,8 кДж
(17.2) С6
Н12
О6
→ 6СО2
+ Н2
О; H
2815,8 кДж
(17.2)
моль
Процесс клеточного дыхания (17.2) является обратной реакцией по отношению к процессу фотосинтеза (17.1). Энергия, выделяющаяся в многостадийном и сложном процессе клеточного дыхания, идет на процессы жизнедеятельности клетки.
Имеются также анаболические дорожки
, на которых затрачивается энергия на построение более сложных молекул из простых. Примером анаболизма является синтез белка из аминокислот (см. Лекцию 6).
Самопроизвольно химические реакции идут только, если свободная энергия Гиббса (зависящая от соотношения величин энтальпии, энтропии и температуры системы) отрицательна (см. Лекцию 13). Если же свободная энергия Гиббса положительна, для осуществления реакции нужен дополнительный приток энергии из окружающей среды. В процессах метаболизма экзотермические реакции (идущие с выделением энергии в окружающую среду) снабжают энергией эндотермические реакции (идущие с поглощением энергии). Любая реакция самопроизвольно достигает состояния равновесия, где беспорядок максимален; чтобы вывести некоторую реакцию из состояния равновесия, клетке требуется дополнительный приток энергии из окружающей среды. Все катаболические реакции, если их проводить в изолированной системе (колбе), самопроизвольно стремятся к термодинамическому равновесию. В живой клетке эти реакции всегда идут в прямом направлении
и не достигают состояния равновесия. Это происходит потому, что продукты одной реакции не накапливаются, а служат исходными веществами для следующей реакции на метаболических дорожках, а отходы жизнедеятельности удаляются из клетки. В результате клетка как биохимическая система все время находится в состоянии, далеком от равновесного, так как выполняется принцип Ле Шателье (см. Лекцию 9).
Клетка «ведет» материю по «метаболическим тропам» с помощью катализаторов –
энзимов
, или ферментов
(
оба термина происходят от греческого и латинского слов, означающих закваска)
– это белковые молекулы-катализаторы, которые обладают свойством избирательно ускорять или замедлять реакции метаболизма. Энзимы снижают энергию активации реакций, не влияя на величину свободной энергии. Если бы все метаболические дорожки открывались одновременно, в клетке царил бы химический хаос. В действительности же, все операции на каждой метаболической дорожке строго регулируются. Дорожки «открываются и закрываются» с помощью управляемого катализа.
Одним из самых обычных механизмов регулирования метаболизма является обратное замедление процессов
. Конечный продукт «запирает» дорожку, служа ингибитором (замедлителем) для соответствующего энзима. Например, клетка использует дорожку из пяти стадий для синтеза аминокислоты под названием изолейцин
из трионина
,
другой аминокислоты. Как только концентрация изолейцина достигает определенной величины, она «выключает» синтез самой себя. Это происходит потому, что изолейцин служит ингибитором того энзима, который служит катализатором самой первой стадии синтеза. Таким образом, клетка не синтезирует больше изолейцина, чем это необходимо.
Сложная структура клетки организует метаболизм во времени и пространстве. Целая «команда» энзимов на нескольких метаболических дорожках объединяется в комплекс. Этот комплекс задаѐт последовательность реакций, в которых продукт первого энзима становится исходным веществом для второго, и так до тех пор, пока не образуется конечный продукт. Для организации процессов метаболизма некоторые энзимы размещаются в определенных местах клетки и даже «ограждаются» мембранами. Даже если энзимы растворяются, они могут быть сильно сконцентрированы вместе со своими субстратами (исходными веществами) в разнообразных метаболических отделах, или органеллах, клетки. Мембраны делят клетку на отсеки, которые имеют свою собственную химическую среду и особую смесь энзимов. Подобно красному, зеленому и желтому сигналам светофора, регулирующим потоки транспорта на дорогах и предохраняющих их от аварий, энзимы поддерживают равновесие между поступлением и расходом вещества, не допуская нехватки или избытка необходимых клетке химических веществ. В целом, метаболизм является механизмом, обеспечивающим высокую упорядоченность и самоорганизацию
клетки как биохимической системы.
17.3. Триплетный код и матричный синтез
Последовательность событий, происходящая между тем моментом, когда клетка делится, и тем, когда делится снова дочерняя клетка, называется жизненным циклом клетки
. Между этими делениями клетка находится в интерфазе,
периоде активного роста и метаболизма. Продолжительность жизненного цикла различных клеток различна в зависимости от их типа и физиологического состояния. Одни клетки делятся каждый час, другим для этого требуется не меньше суток. Некоторые клетки делятся гораздо реже или не делятся совсем: примером служат клетки мышечных тканей и нервные клетки.
Период интерфазы составляет не менее 90% всего жизненного цикла клетки. В период роста и бурной метаболической активности происходит формирование и рост всех составных частей клетки. Затем наступает фаза роста и репликации ДНК и количество хромосом удваивается. Затем наступает вторая фаза роста клетки и ее подготовки к делению. Затем наступает самая драматическая фаза в жизни клетки – митоз
, хорошо различимая в микроскоп. При делении ядра удвоенный набор хромосом распределяется между дочерними клетками, причем каждая получает одинаковое количество генетического материала: по одной молекуле ДНК. Эксперименты показывают, что ошибка в распределении генетического материала между дочерними клетками не превышает величины 1:100000. На последнем этапе митоза между дочерними клетками распределяется цитоплазма.
Репликация ДНК (см. Лекцию 15) является передачей генетической информации
на молекулярном уровне. Как известно, генетическая информация кодируется в молекуле ДНК с помощью четырех нуклеотидов: аденина (А), тимина (Т), гуанина (Г) и цитозина (Ц), располагающихся в определенной последовательности. Основными же рабочими молекулами клетки являются белки, которые состоят из 20 природных, или канонических аминокислот. Набор аминокислот абсолютно одинаков, универсален для всей живой природы. Отличие живых организмов друг от друга состоит в том, в какой именно последовательности расположены аминокислоты в молекулах соответствующих белков, то есть первичной структурой белковых молекул. Аминокислотные последовательности всех белков определяются последовательностью звеньев в одной из двух комплементарных цепочек ДНК. Каким же образом информация, записанная четырехбуквенным алфавитом ДНК, переводится в двадцатибуквенный алфавит белковых молекул?
Решающий вклад в решение этой проблемы внес американский учѐный русского происхождения Георгий Гамов
в 1954 году. Гамов предположил, что образующие первичную структуру белка канонические аминокислоты кодируются сочетаниям трех нуклеотидов, то есть посредством триплетного генетического кода.
Расшифровка этого кода была закончена в 1967 году. Определѐнные тройки нуклеотидов (кодоны
) соответствуют определенным аминокислотам. Так, например, кодон ГЦА соответствует аланину, а ГГЦ – глицину, кодон УУУ – фенилаланину, кодон АУГ – метионину, а кодон ГУГ – валину (СМ. Таблицу 17.1). Так как нуклеотидов существует 4 сорта, а аминокислот – 20 сортов, то всего может существовать 43
= 64 различных кодона. Следовательно, не всякому кодону соответствует аминокислота. Существуют кодоны, выполняющие специальные функции. Они служат как бы стоп-сигналами, обозначающими конец аминокислотной цепи. Однако, таких кодонов немного (УАА, УАГ, УГА). Подавляющее большинство кодонов соответствует какой-либо аминокислоте, причем каждой аминокислоте соответствует не один, а несколько кодонов. То, какой аминокислоте будет соответствовать данный кодон, определяют первые два нуклеотида. Каков третий нуклеотид, не так уж важно, то есть, хотя код и триплетный, главную смысловую нагрузку несет дуплет, стоящий в начале кодона. Иными словами, триплетный код является, по существу, квазидублетным
. Существует правило вырожденности
: если два кодона имеют два одинаковых первых нуклеотида и их третьи нуклеотиды принадлежат к одному классу (пуринов или пиримидинов), то они кодируют одну и ту же аминокислоту.
Специальных терминирующих кодонов не существует. Эту роль в определенных условиях играют кодоны АУГ и ГУГ, обычно отвечающие аминокислотам метионину и валину. Генетический код представлен в таблице 17.1.
Таблица 17.1. Генетический код
| Кодон
|
Кислота
|
Кодон
|
Кислота
|
Кодон
|
Кислота
|
Кодон
|
Кислота
|
| УУУ
|
Phe
|
УЦУ
|
Ser
|
УАУ
|
Tyr
|
УГУ
|
Cys
|
| УУЦ
|
УЦЦ
|
УАЦ
|
УГЦ
|
| УУА
|
Leu
|
УЦА
|
УАА
|
Окончание
|
УГА
|
Окончание
|
| УУГ
|
УЦГ
|
УАГ
|
Окончание
|
УГГ
|
Trp
|
| ЦУУ
|
Leu
|
ЦЦУ
|
Pro
|
ЦАУ
|
His
|
ЦГУ
|
Arg
|
| ЦУЦ
|
ЦЦЦ
|
ЦАЦ
|
ЦГЦ
|
| ЦУА
|
ЦЦА
|
ЦАА
|
Gln
|
ЦГА
|
| ЦУГ
|
ЦЦГ
|
ЦАГ
|
ЦГГ
|
| АУУ
|
Ile
|
АЦУ
|
Thr
|
ААУ
|
Asn
|
АГУ
|
Ser
|
| АУЦ
|
АЦЦ
|
ААЦ
|
АГЦ
|
| АУА
|
АЦА
|
ААА
|
Lys
|
АГА
|
Arg
|
| АУГ
|
Met или
Начало
|
АЦГ
|
ААГ
|
АГГ
|
| ГУУ
|
Val
|
ГЦУ
|
Ala
|
ГАУ
|
Asp
|
ГГУ
|
Gln
|
| ГУЦ
|
ГЦЦ
|
ГАЦ
|
ГГЦ
|
| ГУА
|
ГЦА
|
ГАА
|
Glu
|
ГГА
|
| ГУГ
|
ГЦГ
|
ГАГ
|
ГГГ
|
Где Gly - глицин; Ala - аланин; Val - валин; Leu - лейцин; Ile - изолейцин; Met
- метионин; Phe - фенилаланин; Trp - триптофан; Pro - пролин; Ser - cерин; Thr - треонин; Cys - цистеин; Tyr - тирозин; Asn - аспарагин; Gln - глутамин; Asp - аспарагиновая кислота; Glu - глутаминовая кислота; Lys - лизин; Arg- аргинин; His - гистидин. Кодон АУГ, определяющий аминокислоту метионин, обычно подаѐт сигнал к началу синтеза белка. Терминирующие кодоны УАА, УАГ и УГА подают к завершению синтеза белковой молекулы.
По современным представлениям, ген - это часть молекулы, которая содержит информацию об аминокислотной последовательности одной белковой молекулы
. Как же ген порождает белок? Этот процесс называется матричным синтезом
и происходит в два этапа. На первом этапе, который называется транскрипцией
, специальный фермент узнает последовательность нуклеотидов, расположенную между генами (эту последовательность называют промотором
) и, двигаясь вдоль гена, снимает с него копиюматрицу
в виде молекулы РНК. Молекула РНК сходна по химическому составу с ДНК, однако в отличие от последней, РНК представляет собой одиночную цепочку и содержит не тимин, а урацил. Копирование гена происходит по тому же комплементарному принципу, что и репликация ДНК, только ту роль, которую в ДНК выполняет тимин, в РНК играет урацил. Синтез РНК ведется только по одной из двух комплементарных цепей гена. Энзим, ведущий процесс транскрипции, называется РНКполимеразой. РНК-полимераза снимает с гена, участка длинной молекулы ДНК, копию-матрицу в виде молекулы РНК.
Эта матрица используется на втором этапе синтеза белка, в процессе под названием трансляция
. Именно здесь, на решающем этапе, вступает в силу генетический код. Процесс трансляции очень сложен, в нем принимает участие множество «действующих лиц». Главное из них - рибосома, сложнейший агрегат, построенный из полусотни различных белков и имеющий собственную молекулу РНК, которая в отличие от матричной,
или информационной
, мРНК, называется рибосомной РНК, или рРНК. Рибосома - это своеобразная молекулярная вычислительная машина, переводящая тексты с нуклеотидного языка ДНК и РНК на аминокислотный язык белков, программой для которой служит генетический код. Роль переводчика выполняет РНК еще одного вида - транспортная
РНК. В клетках эукариотов матричная РНК проходит через оболочку ядра, чтобы попасть в цитоплазму, где рибосома синтезирует белок. Сами аминокислоты не могут распознать последовательность кодонов матричной РНК. Именно молекула транспортной РНК отвечает за доставку нужных аминокислот в новую полипептидную цепь. Для этого тРНК должна выполнить две задачи: выбрать нужные аминокислоты и распознать соответствующие кодоны в мРНК. Первая стадия синтеза белка называется инициацией.
Первая аминокислота прикрепляется к транспортной РНК по принципу комплементарности в соответствии с информацией, «записанной» в матричной РНК. В этом процессе участвуют мРНК, тРНК и два участка рибосомы. На второй стадии, который называется стадией наращивания цепи
, к исходной аминокислоте присоединяются друг за другом остальные аминокислоты, пока полипептидная цепь не будет завершена. Заканчивается процесс синтеза белка, когда специальный терминирующий кодон
достигнет определенного участка рибосомы. Готовый белок освобождается и от тРНК, и от рибосомы. В процессе трансляции и после нее белок подвергается нескольким изменениям, которые определяют его трехмерную форму и свойства, отвечающие его функциям в клетке.
17.4. Биохимические процессы в технологической отрасли
Применение биохимических процессов в технологической отрасли особенно перспективно. В живой природе под действием высокоактивных биологических катализаторов – ферментов и гормонов – происходят всевозможные биохимические и каталитические реакции. Они происходят в атмосферных условиях без повышения температуры и давления, с высоким выходом, поэтому подобные технологии, которые можно перенять у живой природы, обладали бы высокой экономической эффективностью. Технологическая биохимия, или биотехнология
, или изучает природные процессы и разрабатывает биохимические методы производства самых разнообразных химических продуктов. Эти методы можно разделить на следующие группы: Биокатализ (ферментация)
В технологической отрасли давно используются биохимические процессы брожения под действием микроорганизмов. Например, в результате брожения глюкозы под действием ферментации дрожжевыми грибками происходит следующая реакция:
 С6
Н12
О6
→ 2СН3
СООН + 2СО2
; H
15,2 кДж С6
Н12
О6
→ 2СН3
СООН + 2СО2
; H
15,2 кДж
моль
Биохимические процессы подобного рода широко применяются в гидролизной промышленности при сбраживании сахаристых веществ с целью получения спиртов и кислот, в виноделии, для производства кормовых дрожжей, в сыроварении, при обработке кож и т.п. Ферментами (или энзимами) называют особые биокатализаторы, которые вырабатываются живыми организмами. По своей химической природе ферменты являются белковыми молекулами, хотя далеко не все белки – ферменты. Молекулы белковых ферментов отличаются своей величиной; их относительная молекулярная масса достигает 100000 единиц относительной атомной массы. По сравнению биокатализаторами, молекулы, на которые действуют ферменты, сравнительно невелики. Эти молекулы называют субстратами
. Небольшие молекулы субстрата располагаются на поверхности фермента, в активных центрах. Продукты реакции уходят с поверхности фермента, а их место занимают новые молекулы субстрата. Общий ход биокаталитического процесса можно представить в виде схемы:
Фермент+субстрат→[ферментно-субстратный комплекс]→фермент+продукты
Многие ферменты проявляют активность и вне клетки, поэтому задача биохимии состоит в том, чтобы провести аналогию между химическим катализом и биокатализом, между ферментами и катализаторами. Достигнуты большие успехи в моделировании биокатализаторов.
Удалось построить модели многих ферментов с высокой активностью и селективностью, но с более простым строением, чем у природных биокатализаторов. Но ни одна полученная модель до сих пор не смогла заменить природную систему, так как такой искусственный катализатор работает всего несколько минут, тогда как в живых организмах ферменты способны самовосстанавливаться и самосовершенствоваться. Ещѐ одним направлением технологической биохимии является биохимия иммобилизированных систем.
Сущность иммобилизации
состоит в закреплении выделенных из живого организма ферментов на твѐрдой поверхности, которая адсорбирует биокатализатор, превращая его в систему, которая может работать непрерывно и стабильно. Биохимический синтез
Биохимический синтез антибиотиков, витаминов, гормонов и прочих ценных биохимических продуктов позволяет удовлетворить потребности населения в этой продукции, так как природных соединений явно не хватает. Особое значение имеют биохимические методы синтеза пищевых продуктов, в частности, белков. Известно, что в мире ощущается недостаток белковых продуктов, и одним из способов увеличения пищевых ресурсов является производство белков биохимическими методами. В настоящее время осуществляется синтез белков в промышленных масштабах из легких масел, нормальных парафинов, метанола, этанола, уксусной кислоты и других органических соединений, получаемых преимущественно из нефти. Используя для биохимического синтеза всего 4 % современной мировой добычи нефти, можно обеспечить белковый рацион 4 миллиардов человек, т. е. почти все население земного шара. В настоящее время установлена первичная структура множества белковых молекул, поэтому синтез белка можно осуществлять химическими методами. Однако это чрезвычайно трудоѐмкий процесс. Например, для синтеза рибонуклеазы
, белка, состоящего из 124 аминокислотных остатков, необходимо провести 369 химических реакций, включающих в себя 11931 стадию. Поэтому всѐ чаще используются синтетические методы получения различных белковых материалов в промышленных масштабах, посредством микробиологического синтеза
ферментными системами микроорганизмов.
Промышленное использование микроорганизмов
С помощью некоторых бактерий, усваивающих водород, можно вовлечь в реакцию кислород и атмосферный диоксид углерода, при этом получить формальдегид и воду. Таким образом, бактерии синтезируют необходимый химической промышленности формальдегид и очищают воздух от двуокиси углерода. Некоторые организмы типа дрожжей способны в соответствующей солевой среде усваивать углеводороды, начиная с метана, и превращать их в белки, которые по аминокислотному составу не уступают самым качественным белкам животного происхождения. Органическая масса дрожжей удваивается за 10-15 минут, поэтому их также можно использовать для производства кормов, поскольку они наполовину состоят из полноценного белка.
Микроорганизмы используются также для извлечения белков и углеводов из травы, древесных и сельскохозяйственных отходов, изготовления искусственной пищи из водорослей (таких, как хлорелла). Одним из методов промышленного использования микроорганизмов является биогеотехнология
, или микробное выщелачивание металлов из руд. Существуют микроорганизмы, способные переводить металлы из рудных минералов в раствор. Механизмы такого выщелачивания бывают разные. Например, некоторые микроорганизмы непосредственно окисляют железосодержащий минерал пирит до соединений трѐхвалентного железа 4FeS2
+ 15O2
+ 2H2
O = 2Fe2
(SO4
)3
+
2H2
SO4,
а ион Fe3+
как сильный окислитель способен перевести в раствор медь из минерала халькоцита Cu2
S + 2Fe2
(SO4
)3
= 2CuSO4
+ 4FeS
O4
+ S или уран из минерала уранита: UO2
+ Fe2
(SO4
)3
= UO2
SO4
+ 2FeSO4
. Реакции окисления являются экзотермическими, при их протекании выделяется энергия, которую микроорганизмы используют в ходе своей жизнедеятельности.
Генно-инженерные технологии
Революция в молекулярной биологии, свершившаяся в результате расшифровки структуры информационной молекулы ДНК, генетического кода и механизма матричного синтеза в молекулярной биологии, привела к появлению в 1970-х годах генной инженерии. Генно-инженерные технологии основаны на рекомбинации молекул ДНК представителей разных видов живых организмов. Наибольшие успехи были достигнуты в области изменения генетического аппарата бактерий. Вводить новые гены в генетический аппарат бактерии можно с помощью небольших кольцеобразных молекул ДНК — плазмид
, присутствующих в бактериальных клетках. В плазмиды «вклеивают» необходимые гены, а затем такие гибридные плазмиды добавляют к культуре бактерий, например кишечной палочки. Некоторые из этих бактерий целиком поглощают такие плазмиды. После этого плазмида начинает работать в клетке как ген, продуцируя в клетке кишечной палочки десятки своих копий, которые обеспечивают матричный синтез новых белков. Историю развития генетической, или генной инженерии можно условно разделить на три этапа. Первый этап связан с доказательством принципиальной возможности получения рекомбинантных молекул ДНК in vitro
(вне организма, в пробирке) и получением гибридов различных плазмид. Была доказана возможность создания рекомбинантных молекул с использованием исходных молекул ДНК из различных видов и штаммов бактерий, их жизнеспособность, стабильность и функционирование. Второй этап связан с началом работ по получению рекомбинантных молекул ДНК между хромосомными генами прокариот и различными плазмидами, доказательством их стабильности и жизнеспособности. Третий этап - начало работ по включению в векторные молекулы ДНК (так называют молекулы ДНК, используемые для переноса генов и способные встраиваться в генетический аппарат клетки-рецепиента) генов эукариот, главным образом, животных. Формальной датой рождения генной инженерии следует считать 1972 год, когда в Стэндфордском университете П. Берг, С. Коэн, Х. Бойлер с сотрудниками создали первую рекомбинантную ДНК, содержавшую фрагменты ДНК вируса SV40, бактериофага и кишечной палочки E. coli. В конце 1970-х годов году в ряде стран (в том числе, в Институте биоорганической химии Академии наук СССР) было осуществлено введение в бактериальную клетку ген, выделенный из человеческого организма. Это достижение позволило решить, в частности, проблему производства ценных лекарственных средств. Наиболее известным примером является синтез инсулина методом генной инженерии. Известно, что у больных сахарным диабетом гормон инсулин, отвечающий за поддержание определѐнной концентрации сахара в крови, не может справляться со своей задачей. Таким больным необходимо постоянно вводить этот гормон извне. На протяжении 60 лет для лечения сахарного диабета применяли говяжий и свиной инсулины, которые по составу отличаются от человеческого на 3 и 1 аминокислоты соответственно, но их активность в организме человека ниже, чем активность человеческого инсулина. Кроме того, инсулин — хотя и небольшой по размерам, но всѐ же белок, и в организме человека со временем накапливаются антитела к нему: организм борется против чужеродных белков, отторгает их. Поэтому бычий или свиной инсулин может начать необратимо нейтрализовываться этими антителами и в результате может исчезнуть прежде, чем успеет оказать лечебное действие. Чтобы этого не произошло, необходимо вводить в организм вещества, предотвращающие этот процесс, но они сами по себе не безразличны для организма. В 1960 году был установлен аминокислотный состав гормона человека и вскоре осуществлен его синтез в лабораторных условиях. Но этот синтез настолько сложен и дорог, что его проводили только в экспериментальных целях, а полученные количества инсулина были недостаточны даже для одной инъекции. В настоящее время для производства человеческого инсулина используют генно-инженерный метод[21]
. При этом ген, ответственный за синтез инсулина, встраивается в ДНК штамма кишечной палочки или дрожжей, которые начинают в больших количествах вырабатывать человеческий инсулин. Преимуществом синтетического человеческого инсулина является полная идентичность естественному гормону человека, поэтому он не обладает иммуногенными свойствами. Человеческий инсулин считают наиболее эффективным средством лечения инсулинзависимого сахарного диабета, поэтому в ряде стран в настоящее время полностью или практически полностью отказались от применения инсулинов животного происхождения. Столь же большой интерес и не меньшее значение имела работа, выполненная в том же институте, по введению методами генной инженерии в бактериальную клетку гена, ответственного за синтез интерферона человека. Интерферон — это белок, играющий исключительно важную роль в борьбе организма против вирусных инфекций. Ген интерферона также был введѐн в клетку кишечной палочки. Созданные штаммы отличались высоким выходом интерферона, обладающего мощным противовирусным действием. Осуществление промышленного производства интерферона является большим достижением, так как предполагают, что интерферон обладает также и противоопухолевой активностью.
Клонирование
В переводе с английского языка клонирование (cloning) означает вегетативное размножение (так размножаются многие растения: отщипнув, например, от куста смородины отросток и посадив его в землю, спустя некоторое время мы получим новое растение). Однако с развитие генной инженерии понятие «клонирование» получило несколько другое значение. Для массового сознания клонирование - это получение неполовым путем организма, точной копии исходной особи. Создание первого удачного клона, шотландской овечки Долли, открыло новые возможности для развития биохимической технологии. Овечка
Долли была создана не для демонстрации возможности создания жизнеспособных клонов высших млекопитающих, а с чисто практической медицинской целью. Генная инженерия открывает широкие возможности превращения домашних животных в «живые лекарства». «Мама» овцы Долли при помощи генной инженерии получила возможность вырабатывать белок, содержащийся в ее молоке, который служит лекарством для больных фиброзом в результате дефекта гена, отвечающего за солевой баланс в легких. Каждый второй такой больной умирает, не дожив до 30 лет. Чтобы сохранить способность вырабатывать такой белок в потомстве уникальной овцы, была проделана уникальная операция: из яйцеклетки другой овцы было удалено ядро, которое заменили ядром соматической (обладающей полным набором хромосом) клетки «мамы» Долли. Яйцеклетка была помещена в матку овцы, которой яйцеклетка и принадлежала, и начала там развиваться. Родившаяся в результате овечка Долли является точной копией не своей биологической матери, а «мамы» Долли, сохранив при этом способность вырабатывать в своем молоке ценный белок-лекарство.
В Технико-фармацевтическом институте Виргинии (США) было получено семейство поросят с «человеческим» геном. От своей прапрабабушки, подвергшейся внедрению этого гена в организм, поросята унаследовали способность синтезировать белок под названием «фактор VIII», который способствует свертыванию крови у больных редкой болезнью, так называемой гемофилией А. Трехсот - шестисот свиноматок, по мнению директора института Билла Веландера, вполне хватило бы, чтобы удовлетворить мировую потребность в лекарстве от гемофилии. Известны также другие клоны (например, полученная российскими учѐными овца Катька, молоко которой способствует подавлению роста раковых опухолей), созданные с целью получения «живых лекарств». ДНК-анализ
Анализ ДНК применяется на практике для идентификации личности в тех случаях, когда невозможно использовать другие способы. Широко известна история идентификации останков, принадлежавших царской семье, расстрелянной в Екатеринбурге в 1918 году. В Англии, в центре судебно-медицинской экспертизы из костной ткани выделили молекулы ДНК и провели их анализ. С точностью до 99,9 % процента было установлено, что останки принадлежат одной семье: отцу, матери и трем их дочерям. Принадлежность семьи к династии Романовых было доказано родством ДНК членов погибшей семьи с ДНК членов английского королевского дома, которые доводятся кровной родней последней русской царице Александре Федоровне.
Каковы же принципы и методика ДНК – анализа? В геноме человека были обнаружены особые участки - мини-сателлиты (крохотные участки молекулы ДНК). В молекулах ДНК разных людей эти участки располагаются в разных местах. Предположим, что исследуются одинаковые участки ДНК двух разных людей, А и Б. На данном участке молекулы ДНК лица А имеется, например, три минисателлита, а на таком же участке ДНК лица Б - два. Эти сегменты ДНК обрабатывают специальными энзимами-рестриктазами, и, в тех местах, где расположены мини-сателлиты, «разрезают» на фрагменты, которые отличаются у А и Б по количеству (два фрагмента у А и один у Б) и по длине. Молекула ДНК, которая, строго говоря, является не кислотой, а солью, в растворе диссоциирует на отрицательно заряженный анион и положительно заряженный катион. Поэтому если поместить раствор, содержащий ДНК, в электролизѐр, то под действием электрического тока анион кислотного остатка ДНК будет двигаться к положительно заряженному электроду. Чем длиннее молекула, тем больше заряд, тем больше сила, увлекающая молекулу к полюсу, то есть тем выше скорость ее движения. Таким образом, в электрическом поле смесь, состоящая из фрагментов ДНК разной длины, распадается на скопления, состоящие из участков ДНК одинаковой длины. Такой метод разделения называется гель-электрофорезом
, разрешающая (разделяющая) способность которого настолько велика, что фрагменты
ДНК, отличающиеся друг от друга всего на одно мономерное звено, четко отделяются друг от друга в виде хорошо различимых полосок. В случае сравнения А и В получились бы две разных «картинки», одна, состоящая из одной полоски, а вторая - из двух. Конечно, реальная картинка будет состоять из сложного сочетания множества полос разной ширины. Если знать какую-нибудь «фамильную» черту, которая передается по наследству (в семье последнего русского царя, например, таким признаком была редкая болезнь гемофилия), то, обнаружив при проведении ДНК-анализа двух образцов ДНК (А и Б) совпадающие минисателлиты соответствующего гена, можно сделать вывод о родстве лиц А и Б.
Нанобиотехнологии
В нанобиотехнологиях
применяются методы и подходы нанотехнологии (см. Лекцию 11) для создания наноустройств для изучения биологических систем. В рамках нанобиотехнологии также изучаются возможности использования для создания наноустройств живых систем; создаются биологические наночипы для диагностики различных заболеваний, в том числе для идентификации возбудителей особо опасных инфекций и токсинов; разрабатываются лекарственные препараты нового поколения и контейнеры в виде наночастиц для адресной доставки лекарственных препаратов в пораженные органы; создаются медицинские нанороботы, способные устранять дефекты в организме больного человека путем управляемых нанохирургических вмешательств, саморазмножающиеся геномы, которые можно применять биотехнологии и медицине для производства лекарств, биосовместимые наноматериалы широкого спектра применения, в том числе для создания искусственных органов.
Из всех имеющихся в наличии биотехнологий в промышленном производстве самое широкое применение находят мембранные технологии (см. Лекцию 18).
17.5. Вопросы и задания
1. Охарактеризуйте основные органеллы живой клетки
2. Каков химический состав клетки?
3. Что такое метаболизм? Какова роль энзимов в метаболизме?
4. Определите возможную последовательность нуклеотидов в молекуле ДНК, которая кодировала бы полипептидную последовательность Phe-Pro-Lys.
5. Составьте схему матричного синтеза.
6. Какие продукты получаются в результате ферментативного брожения глюкозы?
7. Для чего можно использовать микроорганизмы в металлургии?
8. Опишите принцип получения инсулина методом генной инженерии.
9. Что такое клонирование и ДНК-анализ?
10. Каковы возможности нанобиотехнологий?
Лекция 18. Теоретические основы мембранных технологий
Мембранами
называются перегородки, разделяющие различные по составу жидкие или газообразные фазы, и способные, под действием приложенной движущей силы, селективно (раздельно) переносить компоненты разделяемых фаз.
Мембранные технологии
основаны на свойстве мембран действовать как полупроницаемые перегородки
, то есть пропускать сквозь себя одни вещества и не пропускать другие.
Термин «мембрана» для окружающей клетку невидимой плѐнки, служащей барьером между содержимым клетки и внешней средой и, одновременно, полупроницаемой перегородкой, через которую могут проходить вода и некоторые растворенные в ней вещества, был впервые использован немецкими ботаниками фон Молем и К. фон Негели (1817-1891) в 1855 году. В 1877 году ботаник В. Пфеффер (1845-1920) опубликовал свой труд «Исследования осмоса», где предположил существование клеточных мембран, основываясь на сходстве между клетками и осмометрами, имеющими искусственные полупроницаемые мембраны, которые были изготовлены незадолго до этого М. Траубе. Дальнейшее изучение осмотических явлений в растительных клетках датским ботаником X. де Фризом (1848-1935) послужило фундаментом при создании физико-химических теорий осмотического давления и электролитической диссоциации датчанином Я. Вант-Гоффом (1852-1911) и шведским ученым С. Аррениусом (1859-1927). В 1888 году немецкий физхимик В. Нернст (1864-1941) вывел уравнение диффузионного потенциала. В 1890 году немецкий физхимик и философ В. Оствальд (1853-1932) отметил возможность участия мембран в биоэлектрических процессах. В 1895-1902 гг. Э. Овертон (1865-1933) измерил проницаемость клеточной мембраны для большого числа соединений и показал прямую зависимость между способностью этих соединений проникать через мембраны и их растворимостью в липидах (жирах). Это позволяло сделать вывод, что именно липиды формируют плѐнку, через которую проходят в клетку вещества из окружающего раствора. В 1925 году Гортер и Грендел показали, что площадь монослоя липидов, экстрагированных из мембран эритроцитов (клеток крови), в два раза больше суммарной площади эритроцитов. Гортер и Грендел экстрагировали липиды из эритроцитов ацетоном, затем выпаривали раствор на поверхности воды и измеряли площадь образовавшейся мономолекулярной пленки липидов. На основе этих исследований было сделано предположение, что липиды в мембране располагаются в виде бимолекулярного слоя. Вместе с тем имелись экспериментальные данные, которые свидетельствовали о том, что биологическая мембрана содержит в своем составе и белковые молекулы. Эти противоречия экспериментальных результатов были устранены Даниелли и Давсоном, предложившими в 1935 году «бутербродную» модель строения биологических мембран. Согласно этой модели, между фосфолипидными слоями в мембранах располагаются белки.
18.1. Строение биомембран
Клеточная или цитоплазматическая мембрана окружает каждую клетку. Во всех живых клетках биологические мембраны выполняют функцию барьера, отделяющего клетку от окружающей среды, и разделяющего внутренний объем клетки на сравнительно изолированные отсеки. Ядро окружено двумя ядерными мембранами: наружной и внутренней. Все внутриклеточные структуры: рибосомы, митохондрии, эндоплазматическая сеть, аппарат Гольджи, лизосомы представляют собой замкнутые мембранные пузырьки.
Все биомембраны построены одинаково (см. Рис. 18.1); они состоят из двойного слоя липидных молекул (который часто называют липидным бислоем
) толщиной около 6 нм, в которые встроены многочисленные беловые молекулы и молекулярные комплексы.
Рис. 18.1 Строение биомембраны
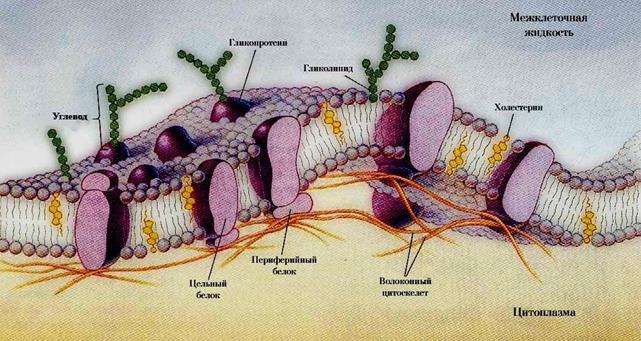
В состав липидов мембран входят в основном фосфолипиды, гликолипиды и холестерин. Некоторые мембраны содержат, кроме того, углеводы, связанные с липидами и белками. Сами по себе, мембраны практически непроницаемы для ионов и полярных молекул, растворимых в воде. Однако одни встроенные в липидный бислой белковые молекулы или комплексы обладают свойствами селективных (т. е. избирательных) каналов для ионов и молекул, а другие служат «насосами», способных активно перекачивать ионы через мембрану. Барьерные свойства мембран и работа мембранных «насосов» приводят к неравновесному распределению ионов между клеткой и внеклеточной средой; это свойство лежит в основе процессов внутриклеточной регуляции и передачи сигналов между клетками в форме электрического импульса.
Белки мембран подразделяются на цельные (интегральные) и периферические. На поверхности цельных белков имеются обширные гидрофобные участки, поэтому эти белки нерастворимы в воде. С липидами мембран они связаны гидрофобными связями и частично погружены в толщу липидного бислоя, а часто пронизывают бислой, оставляя на его поверхности сравнительно небольшие гидрофильные участки. Отделить эти белки от мембраны удается только с помощью таких веществ, которые разрушают липидный слой и переводят белок в растворимую форму. Периферические белки связаны с поверхностью липидного бислоя электростатическими силами, их можно отмыть от мембраны солевыми растворами.
Любая мембрана служит матрицей, на которой располагаются в определенном порядке белки и белковые ансамбли, участвующие в процессах переноса электронов, запасания энергии в форме аденозинтрифосфорной кислоты, регулирования внутриклеточных процессов поступающими извне гормонами и внутриклеточными медиаторами, узнавания других клеток и чужеродных белков, реагирования на свет и механические воздействия и т.д.
18.2. Процессы переноса
Такие низкомолекулярные нейтральные вещества, как газы, вода, аммиак, глицерин и мочевина, свободно диффундируют через биомембраны. Однако с увеличением размера молекулы теряют способность проникать через биомембраны. Например, биомембраны непроницаемы для глюкозы и других сахаров. Проницаемость биомембран зависит также от полярности веществ. Такие неполярные вещества, как бензол, этанол, диэтиловый эфир и многие наркотики, способны легко проходить через биомембраны в результате диффузии. Напротив, для гидрофильных, особенно заряженных, частиц биомембраны непроницаемы. Перенос таких веществ осуществляется специализированными транспортными белками.
Простейшей формой переноса вещества через биомембраны является свободная диффузия (облегченная диффузия). Диффузия часто облегчается определенными мембранными белками, которые можно разделить на две группы:
1. Канальные белки образуют в биомембранах заполненные водой поры, проницаемые для определенных ионов. Например, имеются специфические каналы для ионов Na+
, К+
, Са2+
и Cl-
.
2. В отличие от ионных каналов транспортные белки избирательно связывают молекулы субстрата, перенося их через мембрану. В этом отношении транспортные белки (белки-переносчики) похожи на ферменты. Единственное различие состоит в том, что они «катализируют» направленный перенос, а не какую-либо реакцию. Транспортные белки, или группы транспортных белков, специфичны по отношению к субстратам, подлежащим переносу.
Свободная диффузия и процессы переноса, которые обеспечиваются ионными каналами и переносчиками, осуществляются по градиенту концентрации или градиенту[22]
электрического заряда (или по совместному электрохимическому градиенту). Такие процессы называются «пассивным переносом». Например, по такому механизму в клетки поступает глюкоза из крови, где ее концентрация гораздо выше. В отличие от пассивного переноса, активный перенос идет против градиента концентрации или заряда, поэтому активная транспортировка требует притока дополнительной энергии, которая обычно обеспечивается за счет гидролиза аденозинтрифосфорной кислоты АТФ (см. Рис. 19.2). Некоторые процессы переноса осуществляются за счет световой энергии. Активный перенос может сочетаться с другим, спонтанно идущим транспортным процессом (это так называемый вторичный активный перенос). Так, к примеру, происходит в эпителиальных клетках кишечника и почек, где глюкоза переносится против концентрационного градиента за счет того, что одновременно с глюкозой из кишечника и почек переносятся ионы натрия Na+
. Здесь движущей силой для транспортировки глюкозы является градиент концентрации катионов натрия.
Рис. 18.2. Схема активного переноса: «натриево-калиевый насос»
1.  Транспортный белок мембраны связывает катион натрия Na+,
присутствующий в цитоплазме клетки, что приводит к превращению АТФ в АДФ[23]
с высвобождением фосфатной группы (происходит фосфорилирование белковой молекулы). Транспортный белок мембраны связывает катион натрия Na+,
присутствующий в цитоплазме клетки, что приводит к превращению АТФ в АДФ[23]
с высвобождением фосфатной группы (происходит фосфорилирование белковой молекулы).
2. В результате фосфорилирования белок изменяет свою конфигурацию.
3. Вследствие изменения конфигурации белка ионы натрия выходят за пределы клетки, а транспортный белок связывает катионы калия К+
, находящиеся в межклеточной жидкости.
4. Связывание катионов калия приводит к высвобождению из белковой молекулы фосфатных групп.
5. Потеря фосфатных групп заставляет белок вернуться к первоначальной конфигурации; катионы калия высвобождаются в цитоплазму клетки, и белок вновь начинает связывать катионы натрия.
6. Цикл повторяется.
Эта система активного переноса «накачивает» ионы против градиента концентрации, в результате чего три катиона натрия выводятся из клетки, а два катиона калия внедряются в неѐ из межклеточной жидкости. С помощью подобных транспортных систем осуществляется регулировка ионного состава цитоплазмы, объѐма клеток, величины рН и т.д. Благодаря системам переноса клетки накапливают вещества, необходимые для обеспечения энергетического цикла и метаболических процессов, а также выводят в окружающую среду токсины. Транспортные системы поддерживают ионные градиенты, что имеет большое значение для стимуляции мышечных и нервных клеток. Активный транспорт может идти по механизму унипорта
(облегченной диффузии), когда через биомембрану в одном направлении переносится только одно вещество с помощью канальных или транспортных белков (например, транспорт глюкозы в клетках печени). Активный транспорт может протекать по механизму сопряженного переноса (симпорта)
, когда два вещества переносятся одновременно в одном направлении как, например, транспорт аминокислот или глюкозы вместе с ионами натрия в кишечных эпителиальных клетках, либо в противоположном направлении (антипорт
, или обменная диффузия), как, например, обмен гидрокарбонат-анионов НСО3
-
на хлорид-анионы Cl-
, который происходит в мембранах эритроцитов.
18.3. Современные мембранные материалы и технологии
Применяемые в технологической отрасли современные мембранные материалы можно разделить на следующие виды:
Неорганические: керамические, углеродные, цеолитные, стеклянные и металлические;
Полимерные;
Биомембранные белково-липидные наноструктуры.
Мембранные материалы можно разделить на пористые и непористые. Они могут также различаться по конфигурации: существуют плоские и цилиндрические мембраны, а также мембраны в виде оболочек. На основе различных мембранных материалов разработан широкий ассортимент мембран, мембранных элементов и установок, включая установки для разделения и очистки жидкостей на базе современных неорганических мембран, аппараты для газоразделения, мембранные системы для отделения плазмы крови, мембраны и мембранные элементы для очистки воды и органических жидкостей, мембраны и мембранная аппаратура для современных методов анализа воды и прочие мембранные системы. Важнейшими характеристиками мембран являются производительность
и селективность
. Скорость переноса вещества через мембрану тем выше, чем тоньше мембрана и еѐ активный слой. Толщина современных мембран достигает 100-1000 ангстрем. В селективной мембране скорость переноса одного компонента (или группы компонентов) разделяемой смеси значительно выше, чем скорость переноса других компонентов.
Современные мембраны – это, главным образом, непористые полимерные структуры сложной конфигурации, в которых должны сочетаться такие трудно соединимые качества, как предельно малая толщина рабочего слоя, высокая селективность и механическая прочность. Одним из методов получения таких мембран является метод инверсии
, или разделения фаз
. Полимер растворяют в каком-либо «хорошем» растворителе или смеси растворителей. При медленном испарении растворителя образуется гомогенная непористая мембрана. Если полимерный материал растворѐн в смеси летучего растворителя и осадителя, то при испарении первого возникает тонкий непористый слой, а после начала коагуляции полимера – второй, пористый, слой. Так можно получать асимметричные мембраны. Такие же мембраны можно получать, погружая раствор полимера с образовавшимся на его поверхности тонким селективным слоем в ванну с осадителем. Так, например, изготавливают мембраны из поливинилтриметилсилана
для разделения газов.
Другой способ получения мембран заключается в «бомбардировке» полимерных плѐнок ионами высоких энергий или тяжелыми ионами, которые получают в ускорителях (циклотронах). При последующем протравливании полимерного материала удаѐтся получить систему цилиндрических пор с очень узким распределением по размерам. Полимерные мембраны с очень высокой степенью пористости можно получить при вытяжке частично закристаллизованных полимеров, а также методом выщелачивания специально введенных в полимерные молекулы растворимых добавок (например, неорганических солей). Целый ряд методов получения мембран основан на осаждении тонких полимерных слоѐв на подложках в условиях низкотемпературной плазмы, при межфазной полимеризации, конденсации паров на поверхности подложек тонких слоѐв плѐнок. Все эти методы используют для получения плоских, половолоконных
и композиционных мембран
.
При получении полых волокон исходный раствор полимера продавливают через фильеру особого сечения, а затем, после образования тонкого рабочего слоя на поверхности формирующегося волокна, волокно помещают в коагуляционную ванну, где образуется пористая подложка. Селективный слой при этом можно сформировать как на внутренней, так и на внешней поверхности полого волокна. Обычно внешний диаметр полого волокна не превышает 0,5 мм, а толщина стенки - 10-20 мкм. Половолоконные мембраны имеют гораздо большую, по сравнению с плоскими мембранами, поверхность на единицу объѐма мембранного аппарата. Поэтому предпочтение отдаѐтся мембранам именно такой конфигурации.
Самое широкое применение мембранные технологии находят в процессах очистки промышленных стоков от загрязнений. Мембранные фильтры, применяемые в этой отрасли, можно разделить на следующие группы:
Микрофильтрационные мембраны
с размером пор 0,1 - 1,0 мкм задерживают мелкие взвеси и коллоидные частицы, которые делают воду мутной. Как правило, они используются для грубой очистки или предварительной подготовки воды перед более глубокой очисткой. Ультрафильтрационные мембраны
с размером пор от 0,01 до 0,1 мкм удаляют крупные органические молекулы (молекулярный вес больше 10 000), коллоидные частицы, бактерии и вирусы, не задерживая при этом растворенные соли. Такие мембраны применяются в промышленности и в быту и обеспечивают высокое качество очистки от вышеперечисленных примесей, не изменяя при этом минерального состава воды. Мембраны для ультрафильтрации обычно состоят тонкого рабочего слоя толщиной 0,1-1 мкм, наложенного на крупнопористую подложку толщиной 100 мкм.
Нанофильтрационные мембраны
характеризуются размером пор от 0,001 до 0,01 мкм. Они задерживают органические соединения с молекулярной массой выше 300 и пропускают 15-90 % солей в зависимости от структуры мембраны.
Обратноосмотические мембраны
содержат самые узкие поры и потому являются самыми селективными. Эти непористые или очень мелкопористые мембраны задерживают все бактерии и вирусы, большую часть растворенных солей и органических веществ (в том числе железо и гумусовые соединения, придающие воде цветность, и патогенные вещества). В среднем обратноосмотические мембраны задерживают 97-99 % всех растворенных веществ. Такие мембраны используется во многих отраслях промышленности, где есть необходимость в получении воды высокого качества (розлив воды, производство алкогольных и безалкогольных напитков, пищевая промышленность, фармацевтика, электронная промышленность и т. д.). Обратноосмотические мембраны широко применяются в быту - системы обратного осмоса позволяют получить чистейшую воду, удовлетворяющую российским и европейским стандартам качества для питьевого водопользования, а также всем требованиям для использования в бытовой технике, системе отопления и сантехнике. Мембраны для обратного осмоса имеют асимметричную структуру: они состоят из тонкого селективного рабочего слоя толщиной около 1 мкм, наложенного не крупнопористую подложку толщиной 100-150 мкм. Если подложка изготовлена из другого материала, нежели активный слой, то такие мембраны называются композиционными
.
Принцип мембранных процессов, широко применяемых в очистке воды, состоит в пропускании исходной воды через полупроницаемую мембрану. Под влиянием приложенного давления молекулы воды и некоторые растворенные вещества (размер которых меньше диаметра пор мембраны) проникают через мембрану, тогда как остальные примеси задерживаются. В результате прохождения через мембрану исходная вода разделяется на два потока: фильтрат (очищенная вода) и концентрат (сконцентрированный раствор примесей). Фильтрат подается потребителю, а концентрат сливается в дренаж. Все примеси, размер которых превышает размер пор мембраны, механически не могут проникнуть через мембрану. Благодаря такой технологии, даже при значительном ухудшении параметров исходной воды, качество очищенной воды остается высоким. Мембрана, в отличие от «накопительных» систем очистки воды (активированным углем, ионообменными смолами и т.п.) не накапливает примесей внутри, что исключает вероятность их попадания в очищенную воду. Размер задерживаемых частиц определяется структурой мембраны, то есть размером ее пор.
При переходе от микрофильтрации к обратному осмосу размер пор мембраны уменьшается и, следовательно, уменьшается минимальный размер задерживаемых частиц. При этом, чем меньше размер пор мембраны, тем большее сопротивление она оказывает потоку, и тем большее давление требуется обеспечить для процесса фильтрации. Использование двухступенчатого обратного осмоса (вода дважды пропускается через обратноосмотические мембраны) позволяет получить дистиллированную и деминерализованную воду. Такие системы являются экономически выгодной альтернативой дистилляторам-испарителям и используются на многих производствах (гальваника, электроника и т. д.).
Метод очистки коллоидных растворов и растворов высокомолекулярных веществ от низкомолекулярных примесей посредством мембран называется диализом. Этот метод применяется, например, в аппарате
«искусственная почка».
18.3. Перспективы развития мембранных технологий
Значение мембранной технологии в последние годы резко возросло, прежде всего, как технологии, способной навести мост через пропасть, разделяющую промышленность и экологию. Решением
Правительственной комиссии Российской федерации по научнотехнической политике от 21 июля 1996 года мембранная технология получила статус критической технологии федерального уровня
, так же как катализ, молекулярный дизайн, новые материалы, генная инженерия и другие мировые приоритеты. К основным направлениям развития мембранных технологических процессов относятся следующие:
Разработка мембранных процессов очистки сточных вод с выделением ценных компонентов в машиностроении, целлюлозно-бумажной, текстильной и пищевой промышленности, коммунальном хозяйстве и других отраслях;
Разработка экологически безопасных и ресурсосберегающих технологий для получения ценных нефтепродуктов из природного газа и газового конденсата, отходящих газов нефтепереработки, селективное выделение биологического газа при переработке органических отходов; Переработка вторичного пищевого сырья с выделением ценных компонентов из молочной, сырной и творожной сыворотки, кукурузного и картофельного крахмала, рапса, сои и других пищевых продуктов, очистка пищевых масел от фосфолипидов и следов металлов;
Производство катионопроводящих полимерных мембран для электрохимических генераторов;
Производство мембранных датчиков для компактных
высокочувствительных систем управления и приборов;
Производство мембранных дозаторов лекарственных препаратов с контролируемой скоростью дозировки в ткани и органы, покрытий на раны и ожоги, искусственной поджелудочной железы;
Разработка мембранных технологий для бактериологического контроля качества воды, анализа сыворотки крови, аппаратов для очистки крови; Разработка процессов селективного массопереноса с использованием жидких мембран для извлечения и концентрирования химических продуктов из различных сред;
Разработка теоретических основ получения мембранных катализаторов и мембранных каталитических реакторов, методов исследования проницаемости и дефектности мембранных систем для разделения и концентрирования компонентов;
Производство мембранных реакторов для безотходных процессов получения продуктов при минимальных энергозатратах без сброса сточных вод и выбросов в атмосферу;
Разработка теоретических основ получения новых классов термически и химически стойких мембранообразующих полимеров с функциональными группами разной природы.
Наиболее перспективным направлением является конструирование мембран на молекулярном уровне для переноса «целевых» компонентов (ионов, молекул, коллоидных наночастиц), аналогами которых служат мембраны живых клеток. В этом случае можно добиться очень высокой степени избирательности переноса «целевых компонентов» при общем высоком уровне проницаемости. Такие технологии можно назвать мембранными нанотехнологиями
.
18.4. Вопросы и задания
1. Что такое мембрана?
2. Какое строение имеет мембрана живой клетки?
3. Опишите механизмы переноса вещества через мембрану.
4. Составьте схему действия «натриево-калиевого» насоса.
5. Дайте характеристику современным мембранным материалам.
6. Назовите основные виды мембранных фильтров и дайте им характеристику.
7. Что такое диализ?
8. Каковы перспективы развития мембранных технологий?
Литература
Рекомендуемые учебники по дисциплине «Химия»
1. Н.Л. Глинка. Общая химия - М.: Высшая школа, 2001
2. Н.В. Коровин. Курс общей химии - М.: Высшая школа, 2002
3. Б.Д. Березин, Д.Б. Березин. Курс современной органической химии -
М.: Высшая школа, 2001
4. Основы аналитической химии под ред. Ю.А. Золотова в 2-х томах - М.: Высшая школа, 2001
5. Л.М. Романцева, З.Л. Лещинская, В.А. Суханова. Сборник задач и упражнений по общей химии - М.: Высшая школа, 1991
Литературные источники информации
6. Л.Ю. Аликберова, Е.В. Савинкина, М.Н. Давыдова. Основы строения вещества: методическое пособие-М: МИТХТ, 2004
7. Н.С. Ахметов. Общая и неорганическая химия - М.: Высшая школа, 2002
8. Н. С. Ахметов. Актуальные вопросы курса неорганической химии- М.: Просвещение, 1991
9. З.А. Барсукова. Аналитическая химия: Учебник - М.: Высшая школа,1990
10. Б.Д. Березин, Д.Б. Березин. Курс современной органической химии - М.: Высшая школа, 2001
11. В.П. Васильев. Аналитическая химия: Учебник для вузов в 2-х томах – М., Высшая школа, 2003
12. В.П. Васильев. Сборник задач и упражнений по аналитической химии - М., Высшая школа, 2003
13. Л.М. Витинг, Л.А. Резницкий. Задачи и упражнения по общей химии - М.: Химия, 1995
14. Г.Ф. Воронин. Современная химическая термодинамика, В: Современное естествознание: Энциклопедия в 10 т. - М.: Флинта: Наука, 1999-2000
15. Б. В., Вызов. Природный каучук.- Л.: 1932;
16. Н.Л. Глинка. Общая химия - М.: Высшая школа, 2001
17. Э. Гроссе, Х. Вайсманталь. Химия для любознательных - М.: Химия, 1977
18. Л.Г. Гуськова, И.Л. Шиман. Химия: общие методические указания - М.: Высшая школа, 1978
19. Б. А. Догадкин. Химия эластомеров.- М.: 1972
20. Е.Н. Дорохова, Г.В Прохорова. Аналитическая химия. Физикохимические методы анализа: Учебник – М.: Высшая школа, 1991
21. К.А. Дулицкая, И. В. Кротов, А.Ф. Богоявленский и др. Курс химии в 2-х частях - М., Высшая школа, 1971
22. Е.Н. Еремин Основы химической термодинамики - М..: Высшая школа, 1974
23. О.С. Зайцев. Общая химия - М.: Высшая школа, 1983
24. М.Х. Карапетьянц. Введение в теорию химических процессов - М.: Высшая школа, 1981
25. А.С. Компанеец. Что такое квантовая механика - М.: Наука, 1977
26. Н.В. Коровин. Курс общей химии - М.: Высшая школа, 2002
27. Г.А. Крестов. Теоретические основы неорганической химии - М.: Высшая школа, 1982
28. А.П. Крешков. Основы аналитической химии - М.: Химия,1970
29. Л.Д. Ландау, А.И. Китайгородский. Молекулы - М.: Наука, 1982
30. И.А. Леенсон. Почему и как идут химические реакции - М.: МИРОС,
1994
31. Р.А. Лидин, Л.Ю. Аликберова, Г.П. Логинова. Неорганическая химия в вопросах - М.: Химия, 1981
32. Э.М. Мавсумадзе, Г.А. Аббасова, Т.Г. Захарочкина. Химия в вопросах и ответах с использованием ЭВМ - М.: Высшая школа, 1981
33. Д.И. Менделеев. Основы химии в 2-х томах - М.: Химия, 1947
34. С.П. Муштакова. Колебательные реакции в химии. Соросовский образовательный журнал, 1997, № 7, с. 31–37;
35. В.В. Некрасов. Основы общей химии в 3-х томах – М.: Химия, 1973
36. Г.И. Новиков. Основы общей химии - М.: Высшая школа, 1988
37. Основы аналитической химии под ред. Ю.А. Золотова в 2-х томах -
М.: Высшая школа, 2001
38. Основы аналитической химии: практическое руководство под ред. Ю.А. Золотова - М.: Высшая школа, 2001
39. Н.А. Платэ. Мембранные технологии - авангардное направление развития науки и техники XXI века.
40. М.Э. Полеес, И.Н. Душечкина. Аналитическая химия. – М.: Медицина, 1987
41. А.П. Пурмаль. Как превращаются вещества - М.: Наука, 1984
42. Л.М. Романцева, З.Л. Лещинская, В.А. Суханова. Сборник задач и упражнений по общей химии - М.: Высшая школа, 1991
43. Д.К. Самин. 100 великих научных открытий - М.: Вече, 2000
44. Д.К. Самин. 100 великих ученых - М.: Вече, 2002
45. И.Г. Хомченко. Общая химия - М.: ООО Новая волна, ОНИКС, 2004
46. Химия как экспериментальная наука. Учебное пособие под ред. Д.К
Пиментеля – М.: Мир, 1967
47. Л.А. Шварцман, А.А. Жуховицкий. Начала физической химии для металлургов - М.: Металлургия, 1991
Источники информации в системе Интернет:
http://www.alhimik.ru
http://www.aps.org
http://www.chem.ac.ru
http://www.chem.msu.su/rus
http://chemi.org.ru
http://www.chemport.ru
http://perso.club-internet.fr
http://de.gubkin.ru/chemistry
http://www.educause.edu
http://www.examens.ru
http://him.1september.ru
http://khimiya.narod.ru
http://www.mendeleev.nw.ru
http://www.naukaspb.ru/spravochniki/DEMO_an_chim1/1_1.htm
http://him.1september.ru
http://prometheus/al/ru
http://puggy.symonds.net
http://referat.niv.ru
http://som.fio.ru
http://www.teleport.com
http://www.toehelp.ru
http://www.znanie-sila.ru
http://www.hemi.nsu.ru/ucheb212.htm
http://www.booksite.ru
http://news.ferra.ru/hard/2006/11/03/63307/
http://www.getblob.ru
http://dwb.unl.edu/Teacher/NSF/C10/C10Links/chemistry
http://www.chemnet.ru/rus/publ/Buchachenko/welcome.html#1
http://ru.wikipedia.org/wiki/
http://www.webelements.com/
http://www.cn.ru/edu/chemistry/chemistry/mendl.htm
http://www.sciam.ru/2005/8/lab.shtml
http://www.library.by/shpargalka/belarus/industry/001/ind-001.htm
http://www.fbm.msu.ru/Academics/Manuals/BioPhys/BPh01/membr/01_me04
.html
http://www.biotechnolog.ru/ge/ge1_1.htm
http://www.osmos.ru/index.phtml
http://www.cn.ru/edu/chemistry/chemistry/ucheb1.htm
http://www.physchem.chimfak.rsu.ru/Source/PCC/Termodyn_1.htm
http://www.chemistry.ssu.samara.ru/chem6/hm21.htm
http://schools.techno.ru/doog/bio_kletka/index_04.htm
http://www.krugosvet.ru/about.htm
[1]
Каждый измерительный прибор имеет ограничения, которые определяют его точность, поэтому любые измерения производятся с некоторой ошибкой. Поскольку любая закономерность устанавливается путѐм наблюдений и измерений, то каждый закон, правило или теория включают в себя некоторую неопределѐнность
.
[2]
Решение уравнения Шрѐдингера совпадает с номером периода, в котором находится соответствующий элемент; так, например, у всех элементов II периода, наиболее вероятных местом расположения электронов будут ближайшая к ядру и следующая за ней атомная орбиталь.
[3]
В соответствии с рекомендациями ИЮПАК, относительная атомная масса Аr
(Х) — это молярная масса атома вещества X, отнесенная к 1/12 молярной массы атома углерода-12 (12
С).
[4]
В соответствии с рекомендациями ИЮПАК, эквивалентом называется реальная или условная частица вещества Х, которая в данной кислотно-основной реакции эквивалентна одному иону водорода или в данной окислительно-восстановительной реакции одному электрону.
[5]
В соответствии с рекомендациями ИЮПАК, фактором эквивалентности является число, обозначающее, какая доля реальной частицы вещества Х эквивалентна одному иону водорода в данной кислотно-основной реакции или одному электрону в данной окислительно-восстановительной реакции.
[6]
См. Лекцию 14
[7]
См. Лекцию 9 «Химическое и фазовое равновесие»
[8]
ИЮПАК не рекомендует употреблять термин «процентная концентрация»
[9]
Поскольку в разбавленных растворах активность равна концентрации, в дальнейшем в данном пособии будет использоваться термин «концентрация», хотя следует учесть, что ИЮПАК рекомендует пользоваться термином «активность».
[10]
Энергией активации называют ту минимальную энергию, которой должны обладать молекулы для того, чтобы осуществилось активное соударение, необходимое для распада молекул исходных веществ и образования молекул продуктов реакции.
[11]
В дальнейшем вместо термина «тепловой эффект химической реакции» употребляется термин «энтальпия химической реакции».
[12]
В дальнейшем вместо термина «стандартная теплота образования» употребляется термин «стандартная энтальпия образования».
[13]
На практике абсолютно необратимых реакций не бывает, любая реакция всегда протекает как в прямом, так и в обратном направлении.
[14]
А.М. Жаботинский исследовал механизм реакции Белоусова и научно обосновал открытие последнего.
[15]
См. Рис.11.1
[16]
Свободный радикал – высоко реакционноспособная частица, обладающая неспаренным электроном.
[17]
Энергия Гиббса названа в честь одного из основателей термодинамики, Джозайя Уилларда Гиббса.
[18]
В водном растворе катион водорода H+
внедряется в молекулу воды, образуя катион гидроксония H3
O+
. Существование катиона гидроксония не подтверждено экспериментально, однако допущение о его существовании помогает установить взаимосвязь между кислотой и основанием и поэтому оправданно.
[19]
По современным представлениям, геном является часть молекулы ДНК, хранящая информацию о первичной структуре ОДНОЙ из белковых молекул, из которых построен живой организм.
[20]
ИЮПАК не рекомендует употреблять термин «нормальность», вместо него рекомендуется термин
«молярная концентрация эквивалента» или просто «молярная концентрация» (см. Лекцию 2
«Растворы»)
[21]
В 1978 году исследователи из компании «Генентек» впервые получили инсулин в специально сконструированном штамме кишечной палочки.
[22]
Градиентом называют закономерное количественное изменение, отражающее убывание или возрастание некоего свойства или показателя. Например, градиент концентрации
— это нарастание или уменьшение по какому-либо направлению концентрации растворѐнного вещества, градиент электрического заряда
- увеличение или уменьшение заряда среды по направлению.
[23]
Молекула АТФ состоит из аденина, соединенного с рибозой, которая, в свою очередь, присоединена к цепочке из трех фосфатных групп. Связи между фосфатными группами неустойчивы и при гидролизе разрываются. При этом высвобождается фосфатная группа, а аденозинтрифосфорная кислота АТФ превращается в аденозиндифосфорную кислоту АДФ. Эта реакция является экзотермической и протекает с выделением 59,9 кДж/моль энергии.
|