ТОМСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Химический факультет
Кафедра неорганической химии
СИНТЕЗ БРОМАТОВ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
( Курсовая работа )
Выполнил
Студент 1 курса,
822 группы .
Земляков Д.И.
|
Научный руководитель
К.х.н., доцент
Батырева В.А.
|
Томск 2003
СОДЕРЖАНИЕ.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ НЕОРГАНИЧЕСКОГО СИНТЕЗА 3
ИОННЫЙ ОБМЕН.................................................................. 4
1.Периодическая система и её закономерности как методологическая основа неорганического синтеза................................................................... 9
2.Термодинамический анализ реакций синтеза..................... 9
3.Кинетический анализ реакций синтеза.............................. 11
3. Кинетика и механизм неорганических реакций............... 16
4.Основные методы получения веществ металлов и неметаллов. 20
5.Синтез броматов РЗЭ......................................................... 22
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ...................................... 23
СПИСОК ЛИТЕРАТУРЫ..................................................... 24
Можно выделить три аспекта синтеза: получение известных веществ по известным методикам, получение известных веществ с определенной заданной морфологией (высокодисперсных порошков, монокристаллов, тонких пленок и др.) и получение новых, ранее неизвестных веществ. В учебном практикуме на начальном этапе реальна постановка задачи синтеза известных веществ по известным методикам, и лишь в самых общих чертах возможно ознакомление с проблемой направленного синтеза веществ.
Теоретические основы неорганического синтеза в данном пособии рассматриваются применительно к задачам учебного практикума на базе знаний, полученных студентами при изучении неорганической химии. Это определяет круг включенных в данную главу вопросов и уровень их изложения. Так, в ней рассматриваются методы синтеза, доступные для учебного практикума, и не рассматриваются методы, которые, будучи даже весьма перспективными, в практикуме трудно реализуются, например синтез при высоком давлении, плазмохимический синтез. В главе не приводятся термодинамическая и кинетическая характеристики используемых реакций, за исключением самых общих соображений об их термодинамической возможности и скорости.
Методы неорганического синтеза можно систематизировать, используя разные подходы: по классам синтезируемых соединений (синтез оксидов, гидроксидов, гидридов и т.д.), по типам химических реакций, используемых в синтезе (хлорирование, гидролиз, термолиз и др.), по агрегатному состоянию реагентов (синтез в газовой, твердой, жидкой фазе), по характеру используемой аппаратуры (синтез в вакууме, низкотемпературный синтез и т.д.), по количеству используемых реагентов (макро-, полумикро-, микросинтез). Однако ни одна из этих классификаций не охватывает все разнообразие методов. Например, оксиды металлов чаще всего получают при высокой температуре, а комплексные соединения - в водном растворе. Но эти соединения можно получить и при других условиях, используя самые разные реакции. Так, для получения оксидов металлов можно использовать реакции химического или электрохимического окисления металлов в водном или неводном растворе, окисления их низших оксидов при комнатной температуре и др. При этом синтез можно вести на воздухе и в вакууме, получать вещество в микро-или макроколичестве.
Применительно к задачам практикума по неорганической химии имеет смысл уяснить особенности методов синтеза неорганических соединений разных классов, т.к. это отвечает логике построения теоретического курса. Но согласно логике построения самого практикума, когда ставится задача не только ознакомиться с методами синтеза, но и освоить некоторый объем химического эксперимента, приобрести навыки и умения выполнения определенных химических операций, особый интерес представляет возможность уяснить особенности методов синтеза в разных условиях их проведения. Как обязательная предполагается характеристика особенностей используемых в синтезе химических реакций.
Методы ионного обмена в различных модификациях нашли в настоящее время широчайшее применение не только для аналитических целей, но и в препаративных работах неорганического синтеза. Несмотря на многообразие методов, с применением ионного обмена (распределительная хроматография, хроматография на бумаге, использование жидких ионообменников, тонкослойная хроматография и т. д.) ведущая роль по-прежнему остается за классическими методами ионного обмена.
Успешное решение любой конкретной задачи с применением метода ионного обмена зависит от правильного выбора сорбента и условий его использования. Для этого весьма существенно представлять себе структуру и свойства сорбента как:
химического соединения, так как ионообменная способность, механические и физико-химические свойства сорбентов тесно связаны с их структурой и условиями синтеза.
Ионитами называются органические или неорганические вещества, практически нерастворимые в воде или других растворителях, содержащие активные (ионогенные) группы с подвижными нонами и способные обменивать эти ионы на ионы других электролитов (поглощаемые ионы).
В зависимости от характера введения ионообменных групп все сорбенты делятся на три основных класса:
1. Сорбенты, содержащие в своей структуре кислотные группы, т. е. сорбенты, обладающие свойствами кислот и способные к обмену катионов (катиониты).
2. Сорбенты, содержащие в структуре основные группы, т. е: сорбенты, обладающие свойствами оснований и способные к обмену анионов (аниониты).
3. Амфотерные иониты, т. е. иониты, ионогенная группа которых может вести себя как кислотная или как основная, в зависимости от рН среды.
Существуют также смешанные иониты, т е. сорбенты,. в структуры которых одновременно входят как кислотные, так и основные группы.
Основные требования, предъявляемые к ионообменным смолам, следующие: высокая механическая прочность; химическая устойчивость; минимальная растворимость и небольшая набухаемость при контакте с раствором; высокая обменная способность (смола должна содержать достаточное количество пространственно доступных ионообменных групп); достаточная скорость обмена; желательная избирательность поглощения определенного типа ионов.
Катиониты могут содержать в своем составе различные кислотные группы: сульфогруппу, фосфорнокислые, карбоксильные, фенольные, мышьяково- и селеновокислые и др.
В состав анионитов в качестве функциональных групп могут входить первичные, вторичные и третичные аминогруппы, четвертичные аммониевые и пиридиновые основания.
В зависимости от величины константы диссоциации катионитов в Н+
-форме и анионитов в ОН-
-форме все смолы делятся на сильно- и слабокислотные катиониты и соответственно сильно и слабоосновные аннониты.
При выборе сорбентов в первую очередь нужно учесть, с чем удобнее работать –с катионитом или анионитом. Многие задачи могут быть успешно решены и на том, и на другом типе сорбентов. Например, для разделения ионов металлов можно с успехом применить катиониты. Однако применение для этой же цели анионитов, основанное на разделении анионных комплексов этих металлов, часто бывает проще и быстрее.
Необходимо учитывать также избирательность поглощения сорбентами тех или иных ионов, которая обусловлена химической природой сорбента и определяется относительной прочностью связей обменивающихся ионов в фазе смолы. При этом энергия связи сорбируемого иона зависит не только от прочности связи этого иона с активной труппой сорбента, но и от прочности его связей с любыми другими, так называемыми неактивньгми, структурными группами ионита.
Сильные катиониты и аниониты, например, сульфокатиониты и аниониты типа четвертичных аммониевых оснований, не проявляют большой избирательности в отношении большинства ионов. Большая емкость смол такого типа, а также их способность функционировать в широком интервале рН могут быть использованы для концентрирования сильно разбавленных растворов, для обессоливания и в других случаях, когда необходимо полное извлечение всех катионов или анионов из раствора. Для выделения какого-либо одного элемента из смеси элементов бывает удобно подобрать такой сорбент, который избирательно поглощал бы ионы интересующего элемента.
В настоящее время известно большое количество селективных сорбентов. Синтез таких сорбентов сводится к задаче получения смолы с такой структурой, которая подобна структуре веществ, образующих прочные комплексы или нерастворимые соединения с данным ионом. Так была синтезирована смола (селективно сортирующая никель) путем введения в структуру смолы глиоксимовых группировок.
После выбора соответствующего сорбента необходимо определить область кислотности, в которой работает выбранный ионообменник, и его химическую устойчивость по отношению к тем рабочим средам и температурам, при которых должна проводиться очистка.
Процесс обмена ионов описывается уравнением изотермы, предложенным Б.П.Никольским:
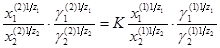
где X1
(2)
и X2
(2)
-концентрации обменивающихся ионов в ионите (ммоль/г); X1
(1)
и X2
(1)
-концентрация обменивающихся ионов в равновесном растворе (ммоль/мл); γ1
(2)
и γ2
(2)
-коэффициенты активности обменивающихся ионов в фазе смолы; γ1
(1)
и γ2
(1)
- коэффициенты активности обменивающихся ионов в фазе раствора; Z1
и Z2
- заряды обменивающихся ионов; K -константа обмена.
Определение коэффициента активности в фазе смолы задача весьма сложная. Однако можно принять, что отношение коэффициентов активности ионов в поглощенном состоянии остается постоянным, и эту величину (можно ввести в константу. тогда, константа ионного обмена:
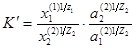
где a1
(1)
и а2
(1)
-активности обменивающихся ионов в равновесном растворе.
Если принять, что интересующий ион отмечен индексом 1, то для этого иона из уравнения никольскова имеем:
 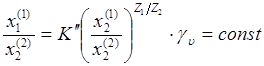
не 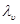 -множитель, содержащий отношение коэффициентов активности обменивающихся ионов в растворе (в соответствующих степенях); -множитель, содержащий отношение коэффициентов активности обменивающихся ионов в растворе (в соответствующих степенях); 
Отношение количества вещества, поглощенного одним грамом сухой смолы, к его концентрации в равновесном растворе называется коэффициентам распределения данного иона. На практике для характеристики поглощения часто определяют именно эту величину, а не константу обмена, которая требует учёта не всегда известных коэффициентов активности.
По определению коэффициент распределения (α)
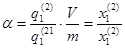
где q1
(2)
и q1
(1)
-содержание исследуемого иона соответственно фазе смолы и в растворе при равновесии, выраженное в любых единицах; V-объем раствора; m-навеска ионнта.
Коэффициент распределения являетcя величиной постоянной не зависит от концентрации интересующего иона в определенном интервале концентраций. Последнее означает, что поглощение ионов элемента -примеси прямо пропорционально его концентрации в растворе, и, следовательно, при очень малых концентрациях изотерма сорбции линейна. Величина коэффициента распределения зависит от природы второго обменивающегося иона, присутствия в растворе других ионов, в том числе мплексообразователей, кислотности раствора, температуры и давления.
Отношение коэффициентов распределения двух различных ионов в одних и тех же условиях называется коэффициентом разделения этих ионов в данных условиях.
Очистку соединений с помощью ионного обмена можно осуществлять разными способами. Если вещества-примеси содержат ионы с величиной заряда, отличающейся от величины заряда очищаемого элемента, то отделение основывается на том, что многозарядные ионы из разбавленных и умеренно концентрированных растворов поглощаются намного сильнее, чем ионы с меньшей величиной заряда. Так, если на сульфокатионитетипа КУ -2 или дауэкс -50 поглотить смесь щелочных, щелочноземельных и редкоземельных элементов, то при элюировании разбавленными растворами хлорной или соляной кислоты в первую очередь будут вымываться ионы щелочных металлов.
В более сложных случаях, когда необходимо разделить элементы, коэффициенты разделения которых близки к единице, чаще всего используют метод комплексообразовательной хроматографии. В этом случае весьма существенными становятся данные о составе, условиях образования и константах устойчивости различных комплексов разделяемых элементов.
В первом варианте все разделяемые ионы сначала поглощаются смолой. Затем производят их (разделение, пропуская (через колонку со смолой раствор комплексообразоаателя., который раздвигает -первоначально образовавшиеся близко расположенные зоны и последовательно вымывает их. При этом подбирают условия, наиболее благоприятные для комплексообравования (рН, температура, концентрация, скорость.пропускания раствора и т. п.). Все ионы вымываются в строго определенном порядке, который задается соотношением прочности связи данного иона со смолой с прочностью образующихся в фильтрате комплексов. Первыми вымываются те ионы, которые образуют наиболее прочные комплексы и слабее всего удержшваютоя смолой.
Во втором варианте комплексообразователь, добавляют к раствору, содержащему смесь разделяемых элементов, и в этом растворе создают условия, благоприятствующие комплексообразованию. Затем производят сорбцию этой смеси комплексов на соответствующем ионите, например, на анионите, если были получены анионные комплексы. При этом лучше всего сорбируютоя наиболее прочные комплексы, которые имеют наибольшее сродство к смоле.
Чем больше различие констант устойчивости, использованных для разделения комплексов, тем полнее и эффективнее достигаемое разделение в обоих вариантах.
Знание констант устойчивости различного вида комплексов очень полезно также при выборе сорбентов, селективно поглощающих определенные ионы. Известно, что во многих случаях сорбированные ионы образуют комплексные соединения со структурными элементами смолы. Очевидно, что чем более
прочные комплексы образуются в фазе смолы, тем большей (избирательностью в отношении данного иона будет обладать смола. В литературе имеется немного работ, посвященных изучению прочности комплексов с функциональными группами :молы. Поэтому, на практике при выборе селективного сорбента пользуются данными об устойчивости аналогичных комплексов в растворах.
Комплексообразование в фазе смолы объясняет, например, высокую избирательность карбоксильных и фосфатных катионов в отношении некоторых катионов. Установлен следующий порядок селективности фосфорнокислых смол в отношении катионов: Th4+
>U 4+
>UO2
2+
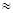 Fe3+
> редкоземельные элементы > Н+
> Сu2+
>Со2+
>Вa2+
>Na+
. Fe3+
> редкоземельные элементы > Н+
> Сu2+
>Со2+
>Вa2+
>Na+
.
Известно также, что многие ионы образуют весьма прочные хелатные комплексы. Оказалось, что смолы, синтезированные (а основе хелатообразующих соединений, обладают весьма высокой избирательностью
по отношению к катионам различных металлов. Поведение хелатных ионитов во многих отношениях сходно с поведением обычных хелатных соединений. В частноси, образование хелатов в фазе ионита сильно зависит от рН и поглощение увеличивается с ростом рН раствора.
Аниониты также обладают способностью координационно связывать некоторые катионы, имеющие ярко выраженную тенденцию к образованию анионных комплексов.
Прежде всего, методологической основой неорганического синтеза являются периодический закон и периодическая система с ее закономерностями (правило об уменьшении стабильности высшей степени окисления с ростом атомного номера в главных подгруппах, диагональное сходство, близость атомных радиусов у атомов элементов пятого и шестого периода за счет f-сжатия, способность элементов к диспропорционированию, полимеризации, комплексообразованию и др.), теории кислотно-основных реакций, теории сольволиза и гидратации, учение о механизмах химических реакций (окислительно-восстановительных, радикальных, обмена лигандов и т.д.), теории химической связи, основные законы химии (при синтезах, например, закон эквивалентов дополняется положением о возможности для многоосновных кислот, многовалентных атомов элементов существования переменного значения кислотно-основного, окислительно-восстановительного эквивалентов).
2
.Термодинамический анализ реакций синтеза
.
Итак, при термодинамической оценке пригодности для синтеза какой-либо обратимой реакции
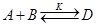
необходимо, чтобы было отрицательным изменение энергии Гиббса реакции
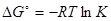
где
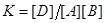
Если для некоторой реакции К >1, то реакцию можно считать практически необратимой. При значении К >1 ожидается достаточно большой выход продуктов, при К <1 реакция должна протекать в основном "справа налево". Условие К <1 не означает, что реакция синтеза продукта не совершается, в этом случае необходимо вычислять равновесный выход продукта, хотя он может быть и мал.
Уравнение, связывающее изменение энергии Гиббса реакции с константой равновесия
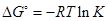
можно записать в виде
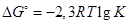
Подставив в это уравнение величины R = 8,З1 ·10-3
кДж/К ·моль и Т= 298 К, получим
∆G°298
= -2,3 8,31 ·10-3
· 298 ·ln K29
8
= -5,70 ln K29
8
.
При значении K ~107
реакция практически проходит до конца в прямом направлении, поэтому значение ∆G° ~ |40| кДж/моль (5,7ּln107
= 5,7ּ7 ~ 40) можно считать в первом приближении границей возможности или невозможности самопроизвольного протекания реакции. Если ∆G° реакции при данной температуре отрицательно и по модулю больше, чем 40 кДж/моль, то такая реакция может протекать в прямом направлении не только при стандартных, но и при любых других условиях. Если же ∆G° реакции при данной температуре положительно и больше 40 кДж/моль, то такая реакция протекать самопроизвольно ни при каких условиях не может.
Если значение ∆G° реакции по абсолютному значению невелико, то при изменении условий процесс может протекать в том или ином направлении. В этом случае для решения вопроса о направлении самопроизвольного протекания реакции недостаточно определить знак и величину ∆G°, нужно рассчитать значение ∆G○
реакции с учетом содержания во взятой смеси исходных и образующихся веществ.
С помощью известных констант равновесия химических процессов можно решить два вопроса: 1) предсказать направление самопроизвольной реакции при заданных условиях эксперимента и 2) при известных исходном составе системы и константе равновесия можно рассчитать равновесный состав смеси, максимально возможный "выход" продуктов, что важно для реакций синтеза. Рассмотрим это на примере расчета газовых реакций.
При решении первого вопроса удобно ввести понятие кажущейся константы равновесия Q как отношения концентраций продуктов к концентрациям реагентов, но не обязательно относящееся к равновесным условиям. Для нахождения Q можно взять отношение, например, начальных концентраций компонентов (C°). Так, для реакции
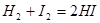
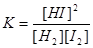 ,
, 
Если имеется больше молекул исходных реагентов, чем должно быть при равновесии, то увеличение знаменателя в выражении для Q приводит к тому, что Q <K, и реакция самопроизвольно пойдет в прямом направлении для образования большего количества продукта; при Q >К самопроизвольно протекает обратная реакция, а при Q =К реагенты и продукты находятся в равновесии.
Определение выхода продуктов реакции проводят с использованием таких понятий, как степень диссоциации α, степень превращения γ, число прореагировавших молей ξ.
Степенью диссоциации α называют долю газа, распавшегося на продукты к данному моменту времени. Значение α можно вычислить для известного значения константы равновесия.
Выразив α через Кр
(константа равновесия) и давление компонентов P , получим :

Выражение степени диссоциации компонентов раствора.
3
.Кинетический анализ реакций синтеза
.
Предметом химической кинетики являются скорости реакций со всеми влияющими на них факторами и интерпретация скорости реакций на основе их механизма. В этом смысле кинетика отличается от термодинамики, в которой рассматриваются начальное и конечное состояния системы вне зависимости от времени протекания этого превращения. Термодинамика обычно рассматривает системы в состоянии равновесия, т. е. в состоянии, в котором скорости прямой и обратной реакций в обратимом процессе равны, что связывает эти две области химии. Однако обратное не верно: скорость реакции нельзя определить только на основе термодинамических данных. Химическую кинетику можно считать более фундаментальной областью науки, но, к сожалению, часто сложность исследуемых процессов делает применение теории химической кинетики довольно трудным.
Рассмотрим равновесие водород -йод в газовой фазе при высокой температуре:
Н2
+ I2
= 2НI
Эта реакция протекает при бимолекулярных столкновениях между молекулами йода и водорода, так что стехиометрическое уравнение в этом случае соответствует истинному механизму взаимодействия. Доказательством правильности такого механизма является то, что скорость образования йодистого водорода, как было показано, пропорциональна как концентрации водорода, так и концентрации йода, т. е.
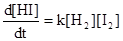
Это, однако, исключительный случай, так как обычно стехиометрическое уравнение не описывает механизма реакции, как, например, для значительно более сложной реакции водорода с бромом, скорость которой, как было показано, определяется уравнением:
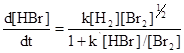
Это можно объяснить следующим механизмом:
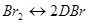
Br + Н2
→ НВr + Н (медленная)
Н + Вr2
→ НВr + Вr (быстрая)
Н + НВr → Н2
+ Вr (быстрая)
Следует отметить, что, зная механизм реакции, не всегда можно дать достаточно определенную интерпретацию экспериментально найденным выражениям для скорости. Иногда с экспериментальными данными согласуются несколько возможных механизмов или вновь полученные данные опровергают ранее принятый механизм.
Очевидно, математическую обработку выражений скоростей реакций через концентрации в определенных степенях
Уравнение:1
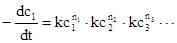
проводить легче, чем для выражений более сложного типа. Только для выражений скорости типа уравнения (1) приемлемо определение порядка реакции n, причем
Уравнение:2
n = n1
+n2
+n3
+……
Из двух рассмотренных выше примеров реакция водорода с йодом -это реакция второго порядка, причем как по водороду, так и по йоду порядок ее равен единице. Понятие порядка реакции неприменимо к взаимодействию водорода с бромом, так как выражение для скорости этого процесса записано не в соответствующей форме.
Если условия проведения реакции таковы, что одна или более концентраций остаются практически постоянными в течение опыта, то эти концентрации можно включить в константу скорости k. В этом случае реакция будет иметь псевдо –n порядок, где n -сумма показателей степеней концентраций, которые в течение эксперимента изменяются. Обычно эти показатели степени -простые положительные числа, но в зависимости от сложности реакции они могут быть дробными или даже отрицательными.
Порядок реакции, определяемый уравнением (2), часто путают с молекулярностью реакции, которая определяется числом молекул, участвующих в элементарном процессе столкновения. Таким образом, молекулярность - это теоретическое понятие, проистекающее из принятого механизма реакции, тогда как порядок - величина эмпирическая; эти две величины могут различаться. Однако бимолекулярные реакции обычно имеют второй порядок, а тримолекулярные реакции -третий порядок, но обратное утверждение не всегда верно. Реакция, которая иллюстрирует только что сказанное, -это окисление ионов Fe2
+
перекисью водорода. Стехиометрическое уравнение ее выглядит так:
2Fr2
+
· aq + Н2
О2
→ 2Fe3
+
· aq + 2OH-
Показано, что выражение для скорости этой реакции
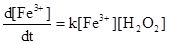
т. е. реакция имеет второй порядок. Схему протекания реакции лучше всего можно представить следующими стадиями:
Fe2
+
· aq + Н2
О2
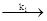 Fe3+ • ао+ОН" +ОН Fe3+ • ао+ОН" +ОН
и
Fe2+
· aq + OH-
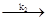 Fe3+
· aq + OH- Fe3+
· aq + OH-
где
k1
= 60 л/моль · сек иk2
= 60 000 л/моль · сек
Так как суммарная реакция состоит из двух последовательных бимолекулярных стадий, то какую -либо молекулярность стехиометрическому уравнению приписать нельзя. Эта схема также иллюстрирует тот факт, что скорость всего процесса определяет самая медленная стадия, так как константа скорости суммарного процесса - это константа скорости первой, более медленной бимолекулярной стадии (т. е. k = k1
). Вторую стадию в этой схеме можно использовать как пример реакции с псевдопорядком.
Для объяснения экспериментальных данных по механизмам реакций широко используют явление изотопного замещения. Так, образец, содержащий радиоактивные ионы Fe2
+
, можно обработать нерадиоактивным образцом, содержащим ионы Fe3
+
, и количество полученных радиоактивных ионов Fe3
+
можно измерить в зависимости от времени. Уравнение Маккея
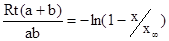
связывает скорость реакции R (т.е. скорость обмена радиоактивностью) с начальными концентрациями a и b реагентов и измеренными радиоактивностями х и 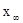 первоначально неактивной формы (в данном случае Fe3
+
) в моменты времени t и первоначально неактивной формы (в данном случае Fe3
+
) в моменты времени t и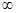 . Поэтому такие реакции являются идеальными для исследования влияния температуры, концентрации и других факторов на скорость реакции. . Поэтому такие реакции являются идеальными для исследования влияния температуры, концентрации и других факторов на скорость реакции.
Таким образом, истинный механизм химических реакций включает мономолекулярные, бимолекулярные или тримолекулярные стадии, по которым реакция идет самопроизвольно при столкновениях между двумя или тремя молекулами. Вероятность одновременного столкновения четырех или более молекул настолько мала, что ею можно пренебречь. Однако можно легко показать, что не все столкновения приводят к химическому взаимодействию. Основными ограничениями, которые лимитируют эффективность столкновений, являются:
а) ориентационные эффекты; очевидно, сложные молекулы могут вступать в реакцию только тогда, когда они соударяются в определенных положениях и в соприкосновение приходят реакционноспособные связи или неподеленные пары электронов. Стерический фактор p показывает, какая часть общего числа соударений приходится на столкновения молекул с такой ориентацией;
б) энергия активации; рассмотрим простую реакцию в газовой фазе
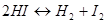
Расстояние H -I в молекуле йодистого водорода равно 1,61 Å и диаметр молекулы равен 3,5 Å.Этот диаметр также должен быть равен расстоянию между двумя атомами водорода или двумя атомами йода в соударяющихся молекулах (удвоенный вандерваальсов радиус; разд. 4.2). Естественно, это расстояние велико по сравнению с расстояниями в молекулах водорода (0,74 Å) и йода (2,67 Å). Следовательно, соударения должны обладать достаточной энергией, чтобы вызвать сжатие молекул НШ, после чего составляющие атомы имели бы возможность подойти друг к другу достаточно близко и вызвать распад этих молекул на водород и йод. Необходимую для этого энергию называют энергией активации реакции, и только те столкновения, которые имеют это минимальное количество энергии, будут эффективными. Часть таких столкновений определяется выражением 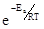 , где Еa
-энергия активации столкновений на один моль. Константа скорости определяется уравнением Аррениуса , где Еa
-энергия активации столкновений на один моль. Константа скорости определяется уравнением Аррениуса

где Z - общее число столкновений между реагирующими молекулами в единичном объеме за одну секунду. Такое простое изложение теории соударений показывает, что она основана на кинетической теории.
Предполагают, что когда взаимодействуют две молекулы, обладающие необходимой энергией активации, то они вначале образуют активированный комплекс, или переходное состояние, который затем разлагается с конечной скоростью с образованием продуктов реакции. Принимают, что скорость реакции определяется скоростью прохождения через переходное состояние, т. е. скоростью прохождения через потенциальный энергетический барьер.Концентрация активированного комплекса в любой момент определяется его равновесием с исходными молекулами. Высота барьера по отношению к энергии исходного состояния равна энергии активации, а разность между энергиями начального и конечного состояний равна теплоте реакции.
С кинетической точки зрения неорганические реакции можно подразделить на две группы:
а) реакции, включающие разрыв и образование ковалентных связей, и
б) реакции, сопровождающиеся простым переносом электронов,
Кроме того, в твердом состоянии реакции протекают еще при перемещении ионов из одной решетки в другую по дефектам решетки. Первый класс реакций можно подразделить на реакции, подобные термическому разложению, рассмотренному ранее, и реакции замещения в координационных соединениях, в которых координированный лиганд замещается другим лигандом из раствора. В общем случае реакции замещения по своему характеру нуклеофильные, так как замещаемый лиганд уносит электронную пару, ранее образовывавшую 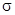 -связь металл -лиганд, а замещающий лиганд приносит пару электронов и поэтому занимает положение с низкой электронной плотностью. По аналогии с органическими соединениями эти процессы обозначаются как SN
-процессы (нуклеофильное замещение). Возможны два основных пути протекания реакции в зависимости от того, происходит ли предварительная диссоциация реагирующего комплекса (мономолекулярный процесс SN
1) -связь металл -лиганд, а замещающий лиганд приносит пару электронов и поэтому занимает положение с низкой электронной плотностью. По аналогии с органическими соединениями эти процессы обозначаются как SN
-процессы (нуклеофильное замещение). Возможны два основных пути протекания реакции в зависимости от того, происходит ли предварительная диссоциация реагирующего комплекса (мономолекулярный процесс SN
1)

или важной стадией является бимолекулярный процесс замещения, скорость которого зависит от концентрации как комплекса, так и замещающего лиганда (SN
2), т. е.
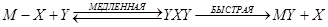
Следовательно, SN
1 -механизм должен привести к активированному комплексу, в котором ион металла имеет меньшее координационное число, чем в исходном комплексе, тогда как SN
2 -механизм требует увеличения числа присоединенных лигандов в переходном состоянии. Необходимо далее рассмотреть разность энергии между реагирующим комплексом и этими переходными состояниями. Если в комплексе нет 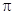 -связей металл -лиганд, то величину скорости реакции можно предсказать, предполагая электростатическое взаимодействие между ионами металла и лигандами. -связей металл -лиганд, то величину скорости реакции можно предсказать, предполагая электростатическое взаимодействие между ионами металла и лигандами.
Наличие двух «вакантных» гране -положении в комплексе с конфигурацией плоского квадрата позволяет предположить, что в этом случае более вероятен SN
2-механизм. В действительности, однако, почти наверное эти транс- положения не будут свободными, и если нет других лигандов, то они будут заняты молекулами растворителя. Эти молекулы растворителя находятся на большем расстоянии, чем лиганды в плоскости квадрата. Поэтому комплекс будет вести себя во многих отношениях так, как если бы он имел конфигурацию плоского квадрата. Две координированные молекулы растворителя очень подвижны и легко могут быть замещены лигандами из раствора. Это облегчает замещение наиболее подвижного лиганда в плоскости квадрата, например
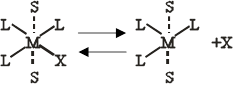 |
Здесь S -молекула растворителя, а рисунок не представляет собой никакой частной стереохимической конфигурации пятикоординационного переходного состояния. К этому переходному состоянию легко присоединяется нуклеофильный реагент У; одновременно комплекс теряет молекулы растворителя и образуется новый комплекс [ML3
Y]. Экспериментально было найдено, что уравнение для скорости реакции типа
[ЭII
X4
] + Y→ [ЭII
X3
Y] + 3
имеет вид
скорость = k1
[комплекс] + k2
[комплекс] [Y]
где k1
-константа скорости реакции первого порядка, которую относят к процессу с SN
2 -механизмом; в этом процессе растворитель -нуклеофильная атакующая единица, k2
-константа скорости реакции второго порядка в процессе с SN
2 -механизмом, в котором нуклеофильной единицей является Y. Если расположить нуклеофильные реагенты в порядке возрастания k1
или k2
то их реакционная способность по отношению к элементу с положительной степенью окисления II будет увеличиваться в ряду:
H2
O ~ OH-
< Cl-
< Br-
~ NH3
~ олефин < ру < NO-
2
< N-
3
< I-
~ SCN-
~ PR3
Очевидно, что по отношению к платине со степенью окисления II большую реакционную способность имеют те лиганды, которые могут быть как σ -донорами, так и π -акцепторами. Платина, занимающая место в конце третьего ряда переходных элементов, имеет несвязанные электроны, необходимые для образования π -связей металл –лиганд. Приведенный выше порядок лигандов определяет также повышение реакционной способности других лигандов, находящихся по отношению к первым в транс -положении. Это явление называют транс -влиянием. Так, в реакции
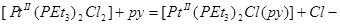
для цис –изомера k1
= 1,7 ·10-2
(при 0°), а для транс -изомера k1
= 10 ·10-6
(при 25°), т. е. когда замещаемый лиганд (Cl-
) находится в транс -положении по отношению к фосфиновой группе, то его замещение идет намного легче, чем когда он находится в транс -положении по отношению ко второму хлору. Можно ожидать, что в комплексах типа 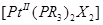 π -связь металл -лиганд прочнее для цис-, чем для транс -изомера. Два атома фосфора в транс -изомере (предполагается, что они лежат вдоль оси х молекулы) могут использовать для образования π -связи только π -связь металл -лиганд прочнее для цис-, чем для транс -изомера. Два атома фосфора в транс -изомере (предполагается, что они лежат вдоль оси х молекулы) могут использовать для образования π -связи только  и и  орбитали, а в цис -изомере орбитали, а в цис -изомере 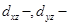 и орбитали. Разность сил связей будет максимальной, когда лиганды Х - слабые π -акцепторы, как в случае иона хлора. Когда же подвижные лиганды не могут действовать как π -акцепторы, их различие в проявлении транс -влияния должно иметь электростатическую природу. Это можно объяснить, предположив поляризуемость подвижного лиганда, влияющего на электронное распределение вокруг центрального атома: этот тип индуктивного эффекта мало проявляется в цис -положении (через 90°), но на лиганд в транс -положении подвижный лиганд оказывает наибольшее влияние. Встречаются такие случаи, когда влияние л-связи и электростатического эффекта само по себе мало, но они дополняют друг друга, приводя к довольно сильному суммарному влиянию на транс -положение; это наблюдается, например, для I-
. Эти предсказания подтверждаются экспериментально исследованием спектров -ядерного магнитного резонанса и определением энергий связей. Так, общая энергия связи цис - и орбитали. Разность сил связей будет максимальной, когда лиганды Х - слабые π -акцепторы, как в случае иона хлора. Когда же подвижные лиганды не могут действовать как π -акцепторы, их различие в проявлении транс -влияния должно иметь электростатическую природу. Это можно объяснить, предположив поляризуемость подвижного лиганда, влияющего на электронное распределение вокруг центрального атома: этот тип индуктивного эффекта мало проявляется в цис -положении (через 90°), но на лиганд в транс -положении подвижный лиганд оказывает наибольшее влияние. Встречаются такие случаи, когда влияние л-связи и электростатического эффекта само по себе мало, но они дополняют друг друга, приводя к довольно сильному суммарному влиянию на транс -положение; это наблюдается, например, для I-
. Эти предсказания подтверждаются экспериментально исследованием спектров -ядерного магнитного резонанса и определением энергий связей. Так, общая энергия связи цис - больше, чем энергия транс -изомера, примерно на 10 ккал. больше, чем энергия транс -изомера, примерно на 10 ккал.
В тетраэдрических комплексах связи металл -лиганд обычно более подвижны, кроме случаев, когда в состав комплекса входят хелатообразующие лиганды. Например, комплекс бериллия с бензоилацетоном можно разделить на оптические антиподы методом избирательной адсорбции одной энантиоморфной формы на колонке с оптически активным кварцем.
Восстановление металлов из оксидов и солеи.
В основе данного метода, охватывающего широкий круг реакций, лежит восстановление выбранного металла какими либо восстановителем в газообразной или жидкой фазе. Реакции в твердой фазе не получили широкого распространения вследствие трудностей, возникающих при разделении продуктов реакции и непрореагировавших исходных веществ.
Восстановителями могут быть вещества в различных агрегатных состояниях, в том числе и твердом состоянии. В последнем случае крайне желательно, чтобы продукты реакции находились в другом агрегатном состоянии (жидком или газообразном). Применение восстановителей в различных агрегатных состояниях. приводит и к различию в кинетических характеристиках системы, что, безусловно, следует учитывать при проведении синтеза.
Чаще всего металлы получают из их оксидов или сульфидов, поскольку, с одной стороны, многие руды представляют собой либо оксиды, либо сульфиды металлов, и, с другой стороны, термодинамика и кинетика подобных реакций достаточно хорошо изучены.
Получение металлов электролизом растворов и расплавов.
Для получения многих металлов в достаточно чистом виде целесообразно применение более сильных восстановителей, нежели водород, оксид углерода, другие металлы и т. д. К числу таких восстановителей относится катод электролитической ячейки, обеспечивающий взаимодействие катиона практически любого металла с электронами.
Все металлы могут быть выделены на катоде при соответствующих условиях. Для разряда какого либо катиона и выделения его на катоде в виде металла требуется приложить к катоду такой потенциал, который преодолел бы стремление ионов ,переходить в, раствор под влиянием присущего каждому из них электрического поля. Поэтому теоретически электролиз возможен только тогда, когда наложенное напряжение превышает собственную электродвижущую силу гальванической пары хотя бы на очень малую величину. Следовательно, низший предел потенциала, необходимого для электроосаждсния катионов, например, из однонормальных растворов, определяется рядом напряжений.
Получение металлов термическим разложением галогенидов и других соединений.
Прочность галогенидов металлов уменьшается от фторидов к йодидам, причем образования некоторых йодидов не превышает десятков единиц кДж/моль. Это позволяет предположить, что при благоприятных условиях возможна диссоциация йодида металла на исходный металл и йод. Аналогично могут вести себя и другие соединения металлов с небольшими отрицательными значениями . Как отмечалось ранее, при диссоциации наблюдается сильное возрастание энтропии (S >0), в то время как энтальпия изменяется незначительно ( близко. к нулю). Поэтому с ростом температуры уменьшается и при некотором значении температуры становится меньше нуля.
Получение неметаллов электролизом растворов и расплавов солей и кислот.
Для данного и последующего параграфов при обсуждении возможности проведения синтеза необходимо руководствоваться значениями окислительных потенциалов химических реакций, приводящих к получению нужных неметаллов. Если при получении металлов путем применения окислительно восстановительных реакций были рассмотрены ряды восстановителей, то при получении неметаллов существенно знание рядов окислителей, могущих привести реакцию к желаемому результату.
Синтез неметаллов в окислительно -восстановительных средах.
Получение галогенидов (кроме фтора), водорода и кислорода в лабораторных условиях чаще осуществляется посредством окислительно -восстановительных реакций в водных растворах, ибо и галогены, и водород, и кислород могут быть получены не только путем анодного окисления, но и при воздействии других более слабых окислителей. Большинство реакций имеют больший окислительный потенциал, нежели реакция перехода молекулярного хлора в хлорид ион, не говоря уже об аналогичных реакциям для брома и йода.
5
.Синтез броматов РЗЭ.
Броматы редкоземельных элементов получают при приливании раствора бромата бария к свежеприготовленному, охлажденному на льду, раствору сернокислой соли. Для ускорения взаимодействия сернокислой соли и бромата бария раствор нагревают. Так полученные броматы не свободны от основных сульфатов. Получение более чистых броматов происходит при сильном встряхивании безводных сульфатов при комнатной температуре с необходимым для растворения количеством воды и твердым броматом бария.
Лучшие результаты в сравнении с указанным выше способом дает перхлоратный метод, который заключается в следующим. Почти нейтральные растворы перхлоратов редко земельных элементов, содержащие до 20% окисей, обрабатывают порошкообразным KBrO3
, кипятят в течение часа и получившийся осадок KClO4
удаляют фильтрованием.
Еще один способ получение броматов РЗЭ, используя катиониты -органические вещества, например смолу, содержащие активную группу. В смолу добавляют насыщенный раствор бромата калия по каплям. В нем делают пробу на ион калия, это может быть реакция с тинитритан калия (желтый осадок). Эту реакцию проводят не на свету, чтобы не произошло разложение свежеприготовленного HBrO3
. Далее полученную кислоту используют для реакции с порошкообразным оксидом редкоземельного элемента.
Пропускаем через ионообменную смолу бромат калия марки «хч» с небольшой скоростью, чтобы ионы Н+
смогли обмениваться с ионами K+
. В результате ионного обмена происходит реакция, указанная ниже:
R-SO3
H + KBr = R-SO3
K + HBrO3
Полученную, таким образом, бромноватую кислоту, приливаем к твердому оксиду иттрия Er2
O3
, причем, оксид РЗЭ берем в избытке. Раствор тщательно перемешиваем стеклянной палочкой до получения нейтральной среды, определяя рН с помощью лакмусового индикатора. Оставляем раствор на несколько часов для выделения избытка оксида иттрия. Аккуратно отделяем полученный маточный раствор от осадка и оставляем его на неделю в естественных условиях ( Т=298 К, Р»105
Па).Декантацию повторяем несколько раз с целью уменьшения количества примесей Er2
O3
в готовом продукте. После вторичной декантации раствор обезвоживают на воздухе при комнатной температуре. В результате образуется кристаллогидраты в виде тонких игл белого цвета общей формулой Er(BrO3
)3
·x9H2
O.
1. Н.А.Скорик. Л.П.Борило. Н.М.Коротченко. Неорганическая химия. Изд-во Томск. гос. ун-та, 1997.- С. 13 - 46.
2. Барнард А. Теоретические основы неорганической химии. – М.:Мир, 1968. - С. 281 – 350.
3. Дей К, Селбин Д. Теоретическая неорганическая химия. – М.:Мир, 1969. – С. 354 –372.
4. Серебренников В.В. Химия редкоземельных элементов. Т.1. – Изд-во Томск. ун-та, Томск, 1959. – С. 235 - 237.
5. Ахметов Н.С. Неорганическая химия. – М.:Высшая школа, 1975.- С. 264 – 286.
|